
京尼平衍生物的合成及其抗类风湿关节炎活性
2022-11-30 03:51柯江涛董心同陈芳园甘珮荣
合成化学 2022年11期
柯江涛, 何 格, 李 志, 董心同, 陈芳园, 甘珮荣, 吴 虹*
(1. 安徽中医药大学药学院,安徽 合肥 230012; 2.新安医学教育部重点实验室 中药研究与开发安徽省重点实验室,安徽 合肥 230012)
类风湿关节炎(Rheumatoid arthritis, RA)是一种慢性炎症性关节疾病,其病理特征为滑膜炎性增生、关节破坏和骨损伤等[1-2]。RA目前无药物可根治,通常需终身治疗以减轻症状[3]。临床上用于治疗RA的药物主要有甾体激素类药物、非甾体抗炎药、病情缓解抗风湿药物和生物制剂等。甲氨蝶呤(Methotrexate, MTX)是目前一线病情缓解抗风湿药物,但其半衰期短,且存在肝毒性、肺炎和肺纤维化等副作用[4]。尽管当前抗RA药物在不断发展,但现有药物均存在较严重的不良反应,故仍需寻找高效低毒的药物治疗RA缓解炎症、改善关节软骨和骨破坏症状。

Scheme 1
GEP为清热解毒中药栀子有效成分栀子苷(Geniposide, GE)水解后的苷元部分,属于环烯醚萜类[5-6]。近年来,已有文献报道证实GE及GEP均具有抗炎、抗过敏及免疫抑制等多种药理活性[7]。GE在体内被肠道β-D-葡萄糖苷酶水解成GEP,后者主要通过抑制NF-κB/IKB-β降解,减少炎症反应过程中前列腺素E2(Prostaglandin, PGE2)和NO的生成,从而显著改善RA患者的症状[8-9]。然而GEP吡喃环上C-3位的羟基和C-10位的羟基极性较大,使其脂溶性降低,难以透过血-关节囊液屏障(Blood-joint barrier, BJB)运输至关节滑膜组织内发挥作用,故其在大鼠体内的生物利用度极低。据此,对GEP进行结构修饰,以提升其脂溶性具有重要意义。
在分子中引入螺环片段可调节化合物的理化性质,同时,相比于开链结构,螺环具有更优的代谢稳定性[10-11]。为此,本文以GEP为先导化合物,并通过在C-10位以螺环为Linker片段引入不同的亲脂性基团改善GEP的脂溶性。为初步验证该设计的合理性,本文以GEP为起始原料,依次经C-3位的选择性乙基化得3-乙基京尼平(2),后者与CDI缩合、2,6-二氮杂螺[3,3]庚烷-2-羧酸叔丁酯半草酸盐成氨基甲酸酯脱Boc得化合物5。化合物5分别与正己酸、环丙酸和苯甲酸缩合成酰胺得化合物6a~6c;与特戊酰氯缩合得目标化合物6d(Scheme 1),其结构经HR-MS(ESI),1H NMR和13C NMR确证。通过CCK-8法测定目标化合物对人类风湿关节炎成纤维滑膜细胞(Human rheumatoid arthritis synovial cells, MH7A)增殖作用的影响。
1 实验部分
1.1 仪器与试剂
ZF-7型三用紫外分析仪; Bruker 400 MH型超导核磁共振仪(DMSO-d6,TMS为内标);Esquire-LC-00075型液质联用质谱仪;MSS型全波长酶标仪; Eppendorf型移液器。
京尼平、乙醇、二氯甲烷(DCM)、三乙胺(TEA)、甲苯、N,N-羰基二咪唑(CDI)、 2,6-二氮杂螺[3.3]庚烷-2-羧酸叔丁酯半草酸盐、N,N-二甲基甲酰胺(DMF)、三氟乙酸(TFA)、1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(EDCI)、 1-羟基苯并三唑(HOBT)、N,N-二异丙基乙胺(DIPEA)、正己酸、环丙烷羧酸、苯甲酸、特戊酰氯,分析纯或化学纯;甲氨蝶呤(MTX)、胎牛血清(FBS,德国Serana公司);DMEM高糖培养基(武汉塞维尔生物科技有限公司);CCK-8检测试剂盒(上海陶术生物科技有限公司)。
1.2 合成
(1) 化合物2的合成
将京尼平1.00 g(4.42 mmol)溶于乙醇(20.00 mL)中,冰浴下加入一滴浓盐酸,置于80 ℃搅拌反应(TLC监测反应进程)。反应完毕,用饱和碳酸氢钠溶液调pH至7~8,依次用水(3×10.00 mL)、饱和食盐水(3×10.00 mL)洗涤,乙酸乙酯(3×20.00 mL)萃取,合并有机相,无水硫酸钠干燥,减压浓缩,残余物经硅胶柱层析[洗脱剂:石油醚 ∶乙酸乙酯=4 ∶1,V∶V]纯化得1.06 g淡黄色油状液体2,产率94.3%, MS(ESI)m/z: 255.1{[M+H]+}。
(2) 化合物3的合成
将化合物21.06 g(4.20 mmol)溶于甲苯(20.00 mL)中,依次加入N,N-羰基二咪唑2.73 g(16.80 mmol)、三乙胺1.34 mL(9.66 mmol),置于55 ℃下搅拌反应(TLC监测反应进程)。反应完毕,在冰浴下缓慢滴加饱和碳酸氢钠溶液溶液淬灭反应至无气泡产生,依次用水(3×10.00 mL)、饱和氯化钠溶液(3×10.00 mL)洗涤,乙酸乙酯 (3×20.00 mL) 萃取,合并有机相,无水硫酸钠干燥,减压浓缩,干燥后直接用于下一步反应,MS(ESI)m/z: 349.1{[M+H]+}。
(3) 化合物4的合成
将化合物31.46 g(4.20 mmol)溶于DMF(20.00 mL)中,依次加入2,6-二氮杂螺[3.3]庚烷-2-羧酸叔丁酯半草酸盐2.45 g(5.04 mmol)和三乙胺2.40 mL(16.80 mmol),室温搅拌反应(TLC监测反应进程)。反应完毕,依次用饱和碳酸氢钠溶液(3×20.00 mL)、水(3×20.00 mL)、饱和食盐水(3×20.00 mL)洗涤,乙酸乙酯 (3×30.00 mL)萃取,合并有机相,无水硫酸钠干燥,减压浓缩,残余物经硅胶柱层析[洗脱剂:石油醚 ∶乙酸乙酯=2 ∶1,V∶V]纯化得1.79 g淡黄色固体4,产率89.0%, MS(ESI)m/z: 479.2{[M+H]+}。
(4) 化合物5的合成
将化合物4692.00 mg(1.45 mmol)溶于二氯甲烷(8.00 mL)中,冰浴下缓慢滴加三氟乙酸(2.00 mL)后移至室温搅拌反应(TLC监测反应进程)。反应完毕,向体系中滴加饱和碳酸氢钠溶液调pH至近中性,依次用水(3×10.00 mL)、饱和食盐水(3×10.00 mL)洗涤、二氯甲烷萃取(3×20.00 mL),合并有机相,无水硫酸钠干燥,减压浓缩,干燥后直接用于下一步反应。
(5)6a~6c的合成
将有机羧酸(2.18 mmol)、 1-(3-二甲氨基丙基)3-乙基碳二亚胺盐酸盐418.00 mg(2.18 mmol)和1-羟基苯并三唑295.00 mg(2.18 mmol)依次溶于二氯甲烷(10.00 mL)中,搅拌下反应1 h;加入化合物5547.40 mg(1.45 mmol)和三乙胺1006.00 μL(7.25 mmol),搅拌反应至终点(TLC监测反应进程)。反应完毕,依次用饱和碳酸氢钠溶液(3×20.00 mL)、水(3×20.00 mL)、饱和食盐水(3×20.00 mL)洗涤,乙酸乙酯(3×30.00 mL)萃取,合并有机相,无水硫酸钠干燥,减压浓缩,残余物经硅胶柱层析[洗脱剂:石油醚 ∶乙酸乙酯=3∶1,V∶V]纯化得目标化合物6a~6c。
((1R, 4aS, 7aS)-1-乙氧基-4-(甲氧羰基)-1,4a,5,7a-四氢环戊烷[c]吡喃-7-基)甲基6-己基-2,6-二氮螺环[3.3]庚烷-2-羧酸酯(6a): 529.00 mg棕色固体,产率76.6%;1H NMR(400 MHz, DMSO-d6)δ: 7.50(s, 1H), 5.78(s, 1H), 4.72~4.61(m, 2H), 4.60~4.49(m, 1H), 4.26~3.91(m, 8H), 3.92~3.80(m, 1H), 3.67~3.55(m, 4H), 3.08~2.97(m, 1H), 2.77~2.66(m, 1H), 2.58~2.50(m, 1H), 2.10~1.97(m, 1H), 1.97(t,J=8.0 Hz, 2H), 1.43(p,J=7.4 Hz, 2H), 1.29~1.17(m, 4H), 1.15(t,J=8.0 Hz, 3H), 0.85(t,J=8.0 Hz, 3H);13C NMR(100 MHz, DMSO-d6)δ: 172.53, 167.36, 155.90, 152.51, 139.36, 128.90, 110.71, 101.22, 65.36, 62.69, 59.86, 57.79, 51.53, 46.19, 38.75, 35.50, 32.39, 31.40, 30.96, 24.29, 22.38, 15.42, 14.31; HR-MS(ESI-TOF)m/z: Calcd for C25H37N2O7{[M+H]+}477.2693, found 477.2698。
((1R, 4aS, 7aS)-1-乙氧基-4-(甲氧羰基)-1,4a,5,7a-四氢环戊烷[c]吡喃-7-基)甲基6-(环丙烷羰基)-2,6-二氮螺环[3.3]庚烷-2-羧酸酯(6b): 538.00 mg淡黄色固体,产率83.0%;1H NMR(400 MHz, DMSO-d6)δ: 7.50(s, 1H), 5.78(s, 1H), 4.71~4.63(m, 2H), 4.55(d,J=16.0 Hz, 1H), 4.36(s, 2H), 4.09(s, 4H), 3.97(s, 2H), 3.91~3.82(m, 1H), 3.66~3.60(m, 4H), 3.07~2.98(m, 1H), 2.77~2.67(m, 1H), 2.58~2.52(m, 1H), 2.11~1.99(m, 1H), 1.52~1.42(m, 1H), 1.16(d,J=8.0 Hz, 3H), 0.71~0.64(m, 4H);13C NMR(100 MHz, DMSO-d6)δ: 172.99, 167.36, 155.92, 152.52, 139.36, 128.92, 110.72, 101.23, 89.35, 65.36, 62.70, 59.78, 58.04, 55.38, 51.53, 46.19, 38.76, 35.51, 32.58, 15.42, 10.03, 7.13; HR-MS(ESI-TOF)m/z: Calcd for C23H31N2O7{[M+H]+}447.2123, found 447.2129。
((1R, 4aS, 7aS)-1-乙氧基-4-(甲氧基羰基)-1,4a,5,7a-四氢环戊烷[c]吡喃-7-基)甲基6-苯甲酰基-2,6-二氮螺环[3.3]庚烷-2-羧酸酯(6c): 505.00 mg淡黄色固体,产率72.1%;1H NMR(400 MHz, DMSO-d6)δ: 7.63~7.57(m, 2H), 7.54~7.41(m, 4H), 5.78(s, 1H), 4.70~4.61(m, 2H), 4.59~4.50(m, 1H), 4.44(s, 2H), 4.19(s, 2H), 4.09(s, 4H), 3.93~3.78(m, 1H), 3.68~3.56(m, 4H), 3.07~2.98(m, 1H), 2.77~2.66(m, 1H), 2.58~2.51(m, 1H), 2.09~2.00(m, 1H), 1.15(d,J=8.0 Hz, 3H);13C NMR(100 MHz, DMSO-d6)δ: 169.24, 167.36, 155.89, 152.52, 139.37, 133.52, 131.45, 128.87, 128.13, 110.72, 101.22, 65.36, 62.97, 62.69, 58.82, 55.39, 51.53, 46.19, 38.75, 35.50, 33.11, 15.42; HR-MS(ESI-TOF)m/z: Calcd for C26H31N2O7{[M+H]+}483.2123, found 483.2130。
(6) ((1R,4aS,7aS)-1-乙氧基-4-(甲氧羰基)-1,4a,5,7a-四氢环戊烷[c]吡喃-7-基)甲基6-新戊酰-2,6-二氮螺环[3.3]庚烷-2-羧酸酯(6d)的合成
将化合物5143.80 mg(0.38 mmol)溶于DMF(4.00 mL)中,依次加入特戊酰氯94.00 μL(0.76 mmol)和DIPEA 182.00 μL(1.03 mmol),室温搅拌反应(TLC监测反应进程)。反应完毕,反应液用乙酸乙酯(3×20.00 mL)萃取,合并有机相,依次用饱和碳酸氢钠溶液(3×10.00 mL)、水(3×10.00 mL)、饱和食盐水(3×10.00 mL)洗涤、无水硫酸钠干燥,减压浓缩,残余物经硅胶柱层析[洗脱剂:石油醚 ∶乙酸乙酯=1 ∶1,V∶V]纯化得112.30 mg淡黄色固体,产率65.0%;1H NMR(400 MHz, DMSO-d6)δ: 7.50(s, 1H), 5.81~5.74(m, 1H), 4.71~4.62(m, 2H), 4.59~4.42(m, 3H), 4.06(s, 5H), 4.00~3.91(m, 2H), 3.93~3.78(m, 1H), 3.69~3.55(m, 3H), 3.06~2.99(m, 1H), 2.76~2.68(m, 1H), 2.57~2.53(m, 1H), 2.09~2.00(m, 1H), 1.15(d,J=8.0 Hz, 3H), 1.08(s, 9H);13C NMR(100 MHz, DMSO-d6)δ: 176.62, 167.36, 155.89, 152.52, 139.38, 128.88, 110.71, 101.23, 65.36, 62.68, 51.54, 46.19, 38.75, 38.37, 35.51, 32.84, 31.62, 30.28, 27.44, 15.43; HR-MS(ESI-TOF)m/z: Calcd for C24H35N2O7{[M+H]+}463.2435, found 463.2441。
1.3 目标化合物体外抗类风湿关节炎活性
(1) 细胞培养
在温度37 ℃下,将人类风湿关节炎成纤维滑膜细胞株MH7A置于含10%胎牛血清的DMEM培养基, 5% CO2的细胞培养箱中培养,用含10 ng/mL的重组人TNF-α刺激MH7A。按1×104个/孔的密度接种于96孔板中,培养24 h,待其完全贴壁后备用。
(2) CCK-8法测定化合物对MH7A的增殖抑制作用
待测京尼平衍生物用DMSO溶解,配置浓度为1×10-2mol/L的溶液,用含5%胎牛血清的DMEM培养基稀释50倍备用。取100 μL稀释好的待测样品加入上述96孔板中,空白对照组加入等量含5%胎牛血清的DMEM培养基溶液,每组设置6个复孔,置37 ℃, 5% CO2条件下培养24 h后,每孔加入10 μL的CCK-8溶液继续避光培养2~4 h,观察孔中颜色由黄变橙后,用酶标仪测定450 nm波长下的吸光度值(OD),以MTX为阳性对照。根据OD值计算化合物对细胞的抑制率,结果见表1。
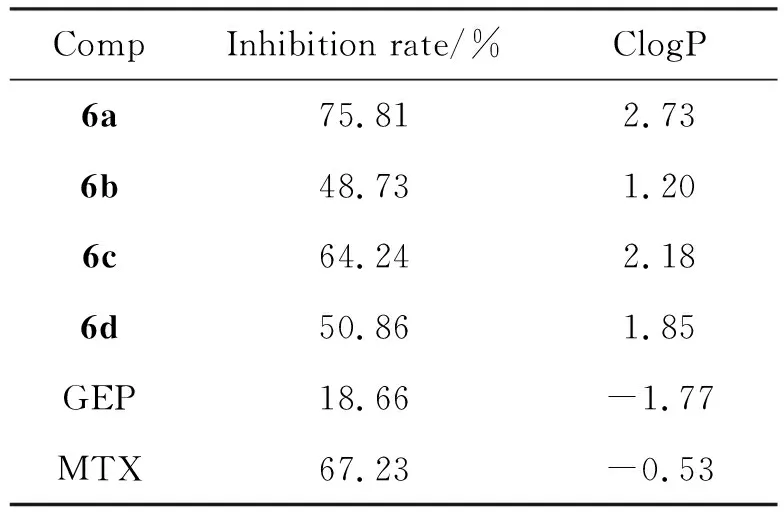
表1 目标化合物对MH7A增殖的抑制活性a
化合物6a~6d在200 μmol/L下对MH7A的增殖抑制率如表1所示。由表1可知,6a对TNF-α诱导的炎性MH7A细胞具有良好的抗增殖活性,其抑制率为75.81%,在相同测试条件下,与阳性药(MTX)抑制活性相当。当其正戊基被替换为环丙基(6b)、苯基(6c)和特戊酰基(6d)时活性下降,抑制率分别为48.73%、 64.24%和50.86%。初步的构效关系研究表明,化合物亲脂性越强,对MH7A细胞的抗增殖活性越强。如前所述,目标化合物对MH7A细胞均具有一定的抗增殖作用,其中化合物6a的抗增殖活性最强,值得进一步优化。
以GEP为先导化合物,通过在C-10位以螺环为Linker片段引入不同的亲脂性基团的方式,设计并合成了4个目标化合物(6a~6d)。目标分子对MH7A的抗增殖活性测试结果表明,6a活性最优,在200 μmol/L下对MH7A的抑制率为75.81%,与MTX相当,具有进一步优化价值。在后续工作中,拟通过丰富螺环Linker与末端脂溶性基团的结构多样性,对该结构类型的目标分子开展深入的构效关系研究,以获得活性更优的化合物用于抗RA生物学评价。
猜你喜欢
工程技术与管理(2022年7期)2022-03-04
陶瓷学报(2021年5期)2021-11-22
科学技术与工程(2021年8期)2021-04-22
哈尔滨工业大学学报(2020年1期)2020-12-21
表面技术(2020年2期)2020-03-04
浙江农业学报(2017年1期)2017-05-17
山东工业技术(2016年15期)2016-12-01
浙江大学学报(工学版)(2016年11期)2016-06-05
文物保护与考古科学(2016年1期)2016-04-16
中国洗涤用品工业(2015年4期)2015-02-28
