
鼠原始生殖细胞报告基因系统的建立及验证
2022-11-25 05:43黄书奇阎晗胡德庆
天津医科大学学报 2022年6期
黄书奇,阎晗,胡德庆
(天津医科大学基础医学院细胞生物学系,天津 300070)
胚胎干细胞(embryonic stem cells,ESCs)是一类具有无限增殖能力、多能性以及自我更新能力的细胞,可以分化成包括生殖细胞在内的多种细胞[1]。在小鼠早期胚胎发育过程中,胚胎基因组在受精卵形成后的2细胞时期开始激活。在8细胞时期,胚胎经历紧束化形成桑椹胚,此时细胞开始出现极性,在胚内的一侧形成一个充满液体的腔,即囊胚腔,在囊胚腔的一侧存在一个小的细胞团,即内细胞团(inner cell mass,ICM),ICM具有分化为成熟个体中全部细胞类型的潜能。随着胚胎的继续发育,ICM将快速增殖并进一步分化,逐步形成3个胚层以及相应的组织和器官[2-5]。哺乳动物胚胎植入子宫后,在BMP信号通路以及Wnt信号通路的共同作用下,近端外胚层细胞分化产生原始生殖细胞(primordial germ cells,PGCs)[6-9],鼠PGCs(mPGCs)呈碱性磷酸酶染色阳性,最早发现于原条后端,存在于胚胎发育的第6.25天(E6.25)。
基因转录调节因子Prdm1(PR/SET Domain 1)的蛋白序列中包含一个N端PR/SET结构域和5个近C端的C2H2锌指结构域(鼠源Prdm1蛋白一级结构示意图见图1)。在早期胚胎发育过程中,Prdm1可以抑制体细胞形成相关信号通路的转导,并致使部分外胚层细胞向生殖细胞谱系分化[10]。Dppa3(developmental pluripotency-associated 3)由母体效应基因编码,主要在PGCs、植入前胚胎和其他多能干细胞中表达,其表达始于小鼠胚胎发育的第7.5天(E7.5),并持续表达至E15.5[11-12]。Dppa3主要包含N端结构域、SAP-like和Splicing-like结构域(包含核定位信号及出核信号)、C端结构域(鼠源Dppa3蛋白一级结构示意图见图1)。卵母细胞中Dppa3的缺失会导致囊胚数量减少、存活幼崽的数量下降[13]。Dppa3特异表达于PGCs和减数分裂前的生殖细胞,在PGCs特化过程中发挥了重要作用。

图1 鼠源Prdm1、鼠源Dppa3蛋白一级结构示意图Fig 1 Schematic diagram of the primary structure of mouse Prdm1 and Dppa3 proteins AB
研究表明,Dppa3与Prdm1在胚胎-胚外界面存在共定位[14-15]。并且,Prdm1和Dppa3为mPGCs的常用标志基因。本研究利用CRISPR基因编辑技术,在小鼠ESCs的Prdm1和Dppa3基因翻译起始位点后分别插入eGFP和mCherry荧光蛋白编码序列,构建了小鼠原始生殖细胞报告基因系统,对于研究小鼠早期胚胎发育过程具有重要意义。
1 材料与方法
1.1 材料
1.1.1 细胞系及质粒mESCs:V6.5购自美国模式培养物集存库(American type culture collection,ATCC)。pSpCas9(BB)-2A-Puro(PX459)、pBluescript SK+质粒购自美国Addgene。
1.2 针对鼠Prdm1和Dppa3基因sgRNA的设计 利用网站(http://crispr.mit.edu)对鼠源Prdm1和Dppa3基因起始密码子位置分别设计一对sgRNA,序列见表1,由生工生物工程(上海)股份有限公司合成。
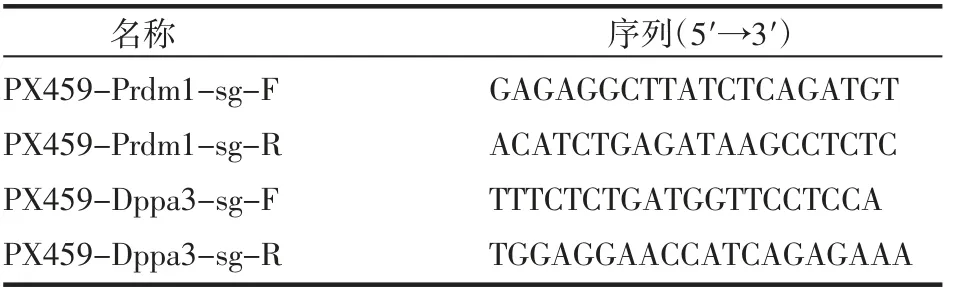
表1 sgRNA名称及序列Tab 1 The name and sequences of sgRNA
1.3 Donor质粒的设计Donor质粒由pBluescript SK+载体骨架以及目的Donor片段构成,Donor片段主要由同源臂序列和要插入的目的片段构成。eGFP-Prdm1 Donor片段由6部分组成,第1部分为切割位点上游5′端500 bp同源臂,第2部分为绿色荧光标签eGFP序列,第3部分为荧光标签与目的基因Prdm1之间的Linker序列,第4部分为切割位点下游Prdm1 1号外显子序列(去除ATG)及450 bp内含子序列,第5部分为FRT序列及Neomycin抗性基因序列,第6部分为3′端500 bp同源臂。mCherry-Dppa3 Donor片段设计原理同上。
1.4 mESC基因组DNA提取(1)收取细胞至1.5 mL EP管中,用200 μL Genomic Lysis Buffer重悬,加入终浓度为200 μg/mL的蛋白酶K,置于金属浴55℃过夜。(2)每管加入200 μL异丙醇,颠倒混匀,置于-20℃静置30 min,后4℃,13 500 r/min离心20 min。(3)弃掉上清,将沉淀用200 μL 75%乙醇洗2次,再用100%乙醇洗1次,弃掉上清,将沉淀晾干。(4)每管加入80 μL ddH2O,吹打混匀,将EP管置于55℃金属浴1 h至沉淀彻底溶解。
1.5 PX459-sgRNA质粒及Donor质粒的构建
1.5.1 PX459-sgRNA质粒的构建(1)引物退火:利用降落PCR,使sgRNA与其互补链形成二聚体。体系:1 00 mmol/L浓度的sgRNA与其互补链各10 μL,ddH2O 30 μL;程序:95℃,5 min;-1℃/min,至25℃;4℃终止。(2)PX459载体线性化:用BbsI内切酶切割载体,体系:PX459载体1 μg;10×FD buffer 2 μL;FastDigestBbsI 0.5 μL;用ddH2O补至20 μL。程序:37℃,1 h。载体酶切产物在电泳后切胶,纯化回收。(3)连接:体系:线性化PX459载体50 ng;双链sgRNA 100 ng,10×T4 ligase buffer 2 μL;T4 ligase 1 μL;用ddH2O补至20 μL。室温连接2 h。(4)转化:将连接产物加入到100 μL DH5α感受态细胞中,冰上静置30 min;42℃水浴热激30 s;冰上静置2 min,加入600 μL LB培养基,37℃,180 r/min摇菌45 min,转化产物涂于Amp抗性的平板,37℃过夜培养,后挑取单克隆菌落,送公司进行测序。
1.5.2 Donor质粒的构建(1)pBluescript SK+载体线性化:用EcoR1和BamH1内切酶切割载体,体系:pBluescript SK+载体1 μg;10×FD buffer 2 μL;Fast-Digest EcoR1和BamH1内切酶各0.5 μL;用ddH2O补至20 μL。程序:37℃,1 h。载体酶切产物在电泳后切胶,纯化回收。(2)eGFP-Prdm1和mCherry-Dppa3 Donor片段扩增:以mESC基因组DNA或质粒为模板,利用PCR扩出Donor片段。体系:5xHF Buffer 10 μL;10 mmol/L dNTP 1 μL;10 μmol/L上下游引物各1 μL;DMSO 1.5 μL;高保真DNA聚合酶0.5 μL;模板:若用质粒为模板,则加入10 ng,若用基因组DNA为模板,则加入250 ng;用ddH2O补至50 μL。程序:98℃预变性3 min;98℃变性30 s,52℃退火20 s,72℃延伸,延伸时间根据片段长度确定,本实验中所使用的高保真DNA聚合酶延伸速率为30 s/kb,扩增循环数为32~35个循环;72℃延伸10 min。(3)连接:体系:线性化pBluescript SK+载体50 ng;Donor片段100 ng;T5 Mix 4 μL,用ddH2O补至20 μL。程序:30℃,40 min。(4)将连接产物转化DH5α感受态中,37℃过夜培养,后挑取单克隆菌落进行菌落PCR鉴定,并送公司进行测序。
1.6 细胞培养及转染mESCs在2i+LIF条件下培养,培养基成分为DMEM、15%FBS、0.1 mmol/L非必需氨基酸、2 mmol/L L-glutamine、1 mmol/L PD、3 mmol/L CHIR、1 000 U/mL LIF。将12孔板用0.1%Gelatin包被30 min,每个孔接种1×105个细胞,每两天传代一次。提取px459-sgRNA、pBluescript-eGFP-Prdm1和pBluescript-mCherry-Dppa3 Donor质粒。将px459-sgRNA和线性化pBluescripteGFP-mPrdm1 Donor质粒各2 μg转染至2×106个细胞中。
1.7 单克隆细胞的筛选及鉴定 转染24 h后更换培养基并加入1.5 μg/mL Puromycin培养48 h,后更换培养基并加入400 μg/mL Neomycin培养1周。挑取单克隆细胞至24孔板中培养,提取基因组DNA进行PCR鉴定。选取阳性细胞,将2 μg pCAGGSFLPe质粒转染至2×106个细胞中,转染24 h后更换培养基并加入1.5 μg/mL Puromycin培养48 h后撤药,继续培养1周后挑取单克隆细胞至24孔板中培养,提取基因组DNA进行PCR鉴定。
1.8 从mESCs向mPGCs的定向诱导 在体外诱导过程中,由mESCs向mPGCs的定向诱导主要由两个步骤组成,第一步是mESCs在ActivinA和bFGF的作用下形成EpiLCs(epiblast-like cells),第二步是在BMP4、BMP8a、SCF、EGF、LIF的作用下由EpiLCs诱导形成PGCLCs(primordial germ cell-like cells)。
EpiLCs诱导:培养基成分为N2B27、1% KSR(Knockout serum replacement)、20 ng/mL ActivinA、12 ng/mL bFGF。将12孔板用0.1% Gelatin包被30 min,每个孔接种1×105个mESCs细胞,培养2 d,每天更换新鲜培养基,得到EpiLCs。
PGCLCs诱导:将2×103个EpiLCs接种到低吸附96孔板的一个孔中,PGCLCs培养基成分为GMEM、15%KSR、0.1 mmol/L非必需氨基酸、2 mmol/L L-glutamine、1 mmol/L sodium pyruvate、55 nmol/L 2-Mercaptoethanol、1 000 U LIF、500 ng/mL BMP4、500 ng/mL BMP8a、100 ng/mL SCF、50 ng/mL EGF。培养4 d后收集细胞,用流式细胞仪检测荧光信号。
2 结果
2.1 CRISPR/Cas9基因敲入系统的构建
2.1.1 PX459-sgRNA质粒的构建结果 对PX459-eGFP-Prdm1-sgRNA和PX459-mCherry-Dppa3-sgRNA重组质粒进行测序,序列比对,编码序列插入的位置和方向均正确,证明质粒构建成功(测序结果见图2)。
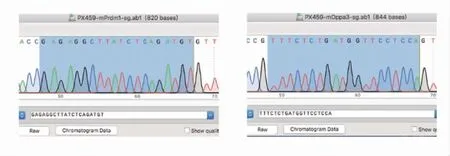
图2 sgRNA测序结果Fig 2 The sequencing results of sgRNA
2.1.2 Donor质粒的构建结果eGFP-Prdm1 Donor片段由6部分组成,第1部分为切割位点上游5′端500 bp同源臂,第2部分为绿色荧光蛋白eGFP编码序列,第3部分为eGFP编码序列与目的基因Prdm1之间的Linker序列,Linker序列为5个连续的甘氨酸,第4部分为切割位点下游Prdm1 1号外显子序列(去除ATG)及450 bp内含子序列,第5部分为FRT序列及Neomycin抗性基因序列,第6部分为3′端500 bp同源臂(图3A)。mCherry-Dppa3 Donor片段同样由6部分组成,第1部分为切割位点上游5′端500 bp同源臂,第2部分为红色荧光标签mCherry序列,第3部分为荧光标签与目的基因Dppa3之间的Linker序列,第4部分为切割位点下游Dppa3 1号外显子序列(去除ATG)及450 bp内含子序列,第5部分为FRT序列及Neomycin抗性编码序列,第6部分为3′端500 bp同源臂(图3B)。以mESCs基因组DNA为模板PCR扩增出Donor片段的第1、4、6部分,从含有相应序列的质粒上PCR扩增出第2、5部分,第3部分Linker序列为公司合成,根据DNA同源重组的原理使用T5酶将这些片段克隆到pBluescript SK+载体上,经公司测序,序列插入的位置和方向均正确,Donor质粒构建成功。
2.2 鼠Prdm1基因翻译起始位点后插入eGFP编码序列的细胞株鉴定 引物设计方案见图3A。提取不同单克隆细胞的基因组DNA进行PCR,首先通过引物P3和P4从37个单克隆中鉴定出2个阳性克隆,以野生型(WT)细胞为对照,4号和37号克隆在750 bp位置出现条带,说明这2个克隆均成功插入eGFP编码序列。接下来通过引物P1和P2判断这2个阳性克隆是纯合子或是杂合子,其中WT在250 bp处出现条带,4号克隆只在1 kb处出现条带,为纯合子;37号克隆分别在250 bp和1 kb处出现条带,为杂合子。为了进一步确认eGFP编码序列是否正确插入到鼠Prdm1基因翻译起始位点后,从5′同源臂上游选取上游引物P7,在eGFP编码片段上选取下游引物P8,PCR结果显示,4号克隆和37号克隆均在700 bp位置出现条带,说明eGFP编码序列正确插入到鼠Prdm1基因翻译起始位点后。
为了提高基因编辑的效率,在Prdm1的1号和2号外显子之间的内含子上插入了Neomycin抗性编码序列,其表达产物可以给细胞提供复制压力,在使用Neomycin进行筛选时,细胞为了存活,会产生更多的同源重组,同时在Neomycin抗性编码片段两端插入了同向的FRT位点,外源表达的重组酶Flippase可以切除两个方向相同的FRT位点之间的序列,从而达到去除Neomycin抗性编码片段的目的。挑选4号和37号阳性克隆,转入pCAGGS-FLPe质粒后用药物进行筛选,提取不同细胞克隆的基因组DNA进行PCR,引物设计方案见图3A。用引物P5和P6进行PCR,成功去除Neomycin抗性编码片段的克隆无法扩出300 bp的条带(图4A)。
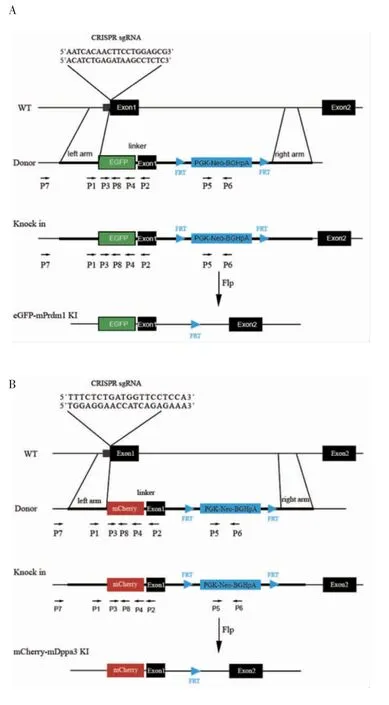
图3 CRISPR介导的基因敲入策略模式图Fig 3 Schematic diagram of CRISPR-mediated gene knock-in strategy
2.3 鼠Dppa3基因翻译起始位点后插入mCherry编码序列的细胞株鉴定 分别向上述4号和37号细胞克隆转染PX459-mCherry-Dppa3-sgRNA和mCherry-Dppa3-Donor质粒,进行抗性筛选后挑取单克隆并提取基因组DNA进行鉴定,基因型鉴定引物设计方案见图3B。挑取4号克隆中的纯合子以及37号克隆中的杂合子,转染pCAGGS-FLPe质粒去除Neomycin抗性基因片段。最后得到4-14号克隆为eGFP-Prdm1/mCherry-Dppa3双纯合子细胞,37-8号克隆为eGFP-Prdm1/mCherry-Dppa3双杂合子细胞(图4B)。
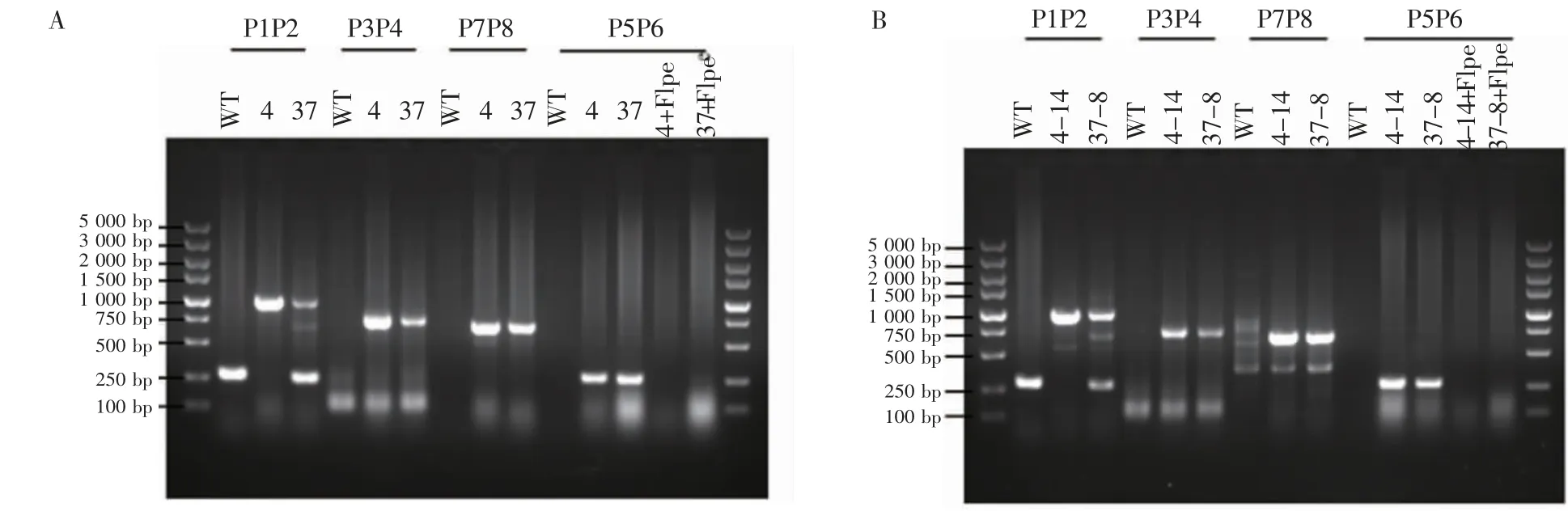
图4 eGFP-Prdm1/mCherry-Dppa3KnockIn细胞基因型鉴定结果Fig 4 The genotyping results of eGFP-Prdm1/mCherry-Dppa3 Knock In cells
2.4 流式检测PGCs诱导结果 在进行体外诱导前,即2i+LIF培养条件下,对WT细胞、eGFPPrdm1/mCherry-Dppa3双杂合子细胞及双纯合子细胞进行流式分析,结果见图5,WT细胞中未检测到eGFP和mCherry荧光信号,Knock In细胞中可以检测到微弱mCherry和eGFP荧光信号,并且纯合子的mCherry和eGFP荧光信号比杂合子强,此结果符合在mESCs中Dppa3基因有少量表达,Prdm1基因也有微弱表达。
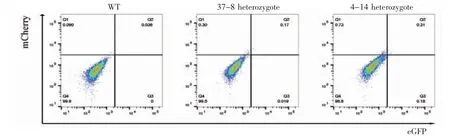
图5 未诱导时WT及Knock In细胞流式检测结果Fig 5 Flow cytometry results of WT and Knock In cells without induction
选用WT细胞和eGFP-Prdm1/mCherry-Dppa3 Knock In细胞进行PGCs诱导实验(实验流程示意图见图6A),使用Activin A、bFGF和1%KSR刺激2 d后,细胞形态从克隆状变成扁平的上皮样,说明成功将ESCs诱导为EpiLCs;然后将3×103个细胞转移到低吸附96孔板中培养,加入500 ng/mL BMP4、500 ng/mL BMP8a、100 ng/mL SCF、50 ng/mL EGF、1 000 U LIF,培养4 d后收集细胞进行流式分析,培养过程见图6B,流式分析结果见图7,与WT相比,Knock In细胞出现明显的eGFP和mCherry阳性,成功由EpiLCs状态诱导为PGCLCs,成功构建了eGFP-Prdm1/mCherry-Dppa3 Knock In细胞系。
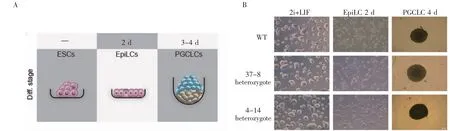
图6 PGCLCs诱导实验流程示意图Fig 6 Schematic diagram of PGCs induction experiment
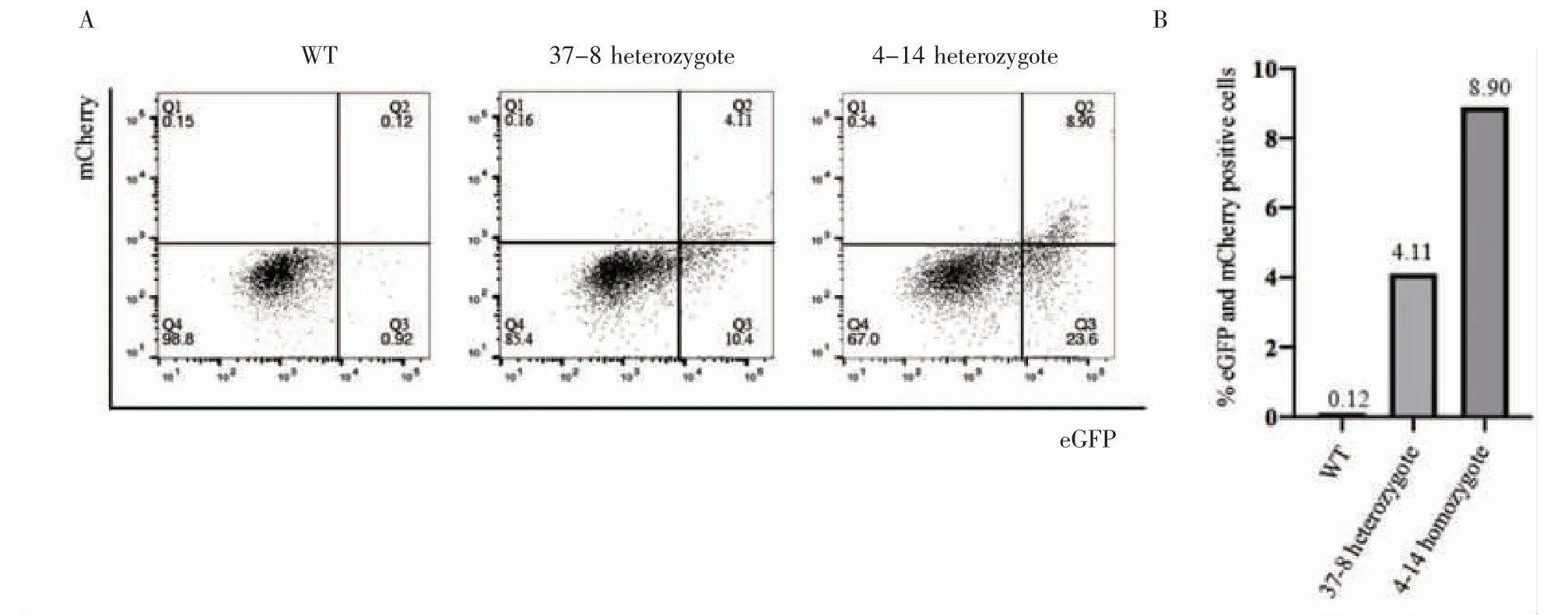
图7 原始生殖细胞诱导流式检测结果Fig 7 The flow cytometry results of primordial germ cell induction
3 讨论
PGCs是生殖细胞的起源,在早期胚胎发育过程中,PGCs在尿囊底部形成,随着胚胎发育,大量的PGCs沿着后肠迁移到生殖嵴,最终分化为功能性配子,这种早期谱系选择依赖于BMP/Smad信号通路激活Prdm1基因表达,从PGCs形成直至迁移到生殖嵴,Prdm1一直持续表达[16]。在小鼠中,Prdm1基因缺失可导致胚胎出现大量的血液渗漏和组织凋亡,并在妊娠中期死亡。Prdm1纯合子突变体胚胎无法产生PGCs,杂合子突变体胚胎中PGCs数量显著减少[17]。Dppa3最初是在小鼠原肠胚时期出现并一直持续表达到E15.5。Dppa3缺陷的雌性个体可以正常受精,但由于染色质压缩和基因转录抑制,很难产生后代[18]。在生殖细胞发育过程中,PGCs在不同的时间点表达不同的基因并进行特定的表观遗传重塑。当在小鼠胚胎成纤维细胞中过表达3种特定的生殖细胞基因(Dppa3、Oct4和Nanos2)时,许多与干细胞自我更新以及生殖细胞程序相关的基因会被激活[19],这些都表明Dppa3在生殖细胞发育过程中具有重要作用。
胚胎发育以及相关的遗传信息通过配子传递给后代,因此研究PGCs的特化过程对于理解早期胚胎发育过程以及相关疾病的机制具有重要意义。但是早期胚胎发育过程中,体内PGCs数量十分稀少,这极大影响了对于早期生殖细胞发育进程及相关分子机制的探究,因此需要在体外建立由ESCs分化形成PGCs的培养系统。在之前的研究中,Ohinata等[20]构建Prdm1-Venus及ECFP-Dppa3 BAC(Bacterial artificial chromosome),用显微注射针将线性化的外源DNA片段注入小鼠受精卵的原核中,从而获得Prdm1-Venus及ECFP-Dppa3转基因小鼠,将这两种小鼠进行后续交配以获得双纯合小鼠(将双纯合命名为BVSC:Blimp1-Venus and Stella-ECFP),并从与BVSC雄性交配的雌性小鼠囊胚中获得带有PGC报告基因的ES细胞系。此方法中外源DNA为随机整合,不同整合位点的PGC报告基因的表达水平有很大的差异需要通过建系筛选外源基因高表达的系,过程繁琐,并且外源DNA的随机整合还可能影响内源基因的表达。此外,真核细胞中的很多顺式作用元件与靶基因相距较远,BAC质粒中可能缺失这些远程调控元件,因此利用BAC质粒获得的带有PGC报告基因的ES细胞系并不一定能真实地反映PGC基因在ES细胞分化过程中的表达。CRISPR-Cas9系统通过非常短小的guideRNA形成靶向特异性,该系统极大地简化了基因组编辑操作,应用领域涵盖干细胞工程、基因治疗、组织和动物疾病建模等诸多方面。
在本实验中,利用CRISPR技术实现基因组定点编辑,在mESCs中构建mPGCs分化的报告基因系统,为PGCs提供稳定、可靠和安全的标记方法,有助于后续进一步探究PGCs分化的分子机制,也可促进对早期胚胎发育过程的理解以及不孕不育等相关疾病的研究。
猜你喜欢
环球时报(2022-09-20)2022-09-20
中国生殖健康(2020年5期)2021-01-18
今日农业(2020年24期)2020-12-15
国际放射医学核医学杂志(2020年4期)2020-07-27
中国医学影像技术(2019年10期)2019-10-24
中国生殖健康(2018年5期)2018-11-06
精准医学杂志(2018年5期)2018-02-12
临床与实验病理学杂志(2017年6期)2017-03-07
新疆医科大学学报(2015年10期)2015-12-26
小资CHIC!ELEGANCE(2015年15期)2015-09-01
