
GC-MS/MS测定地表水中32种农药残留
2022-11-25 12:18黄碧嘉何秀琴罗绮雯
粮油食品科技 2022年6期
赵 悦,黄碧嘉,何秀琴,罗绮雯
(1. 拱北海关技术中心,广东 珠海 519000;2. 中山海关技术中心,广东 中山 528400)
农药残留是指农药使用后一定时期内没有被分解而残留于生物体、收获物、土壤、水体、大气中的微量农药原体、有毒代谢物、降解物和杂质的总称[1]。作为农业大国,合理的使用农药,有助于我国的农业生产,保障我们的餐桌安全。但在实际生产中,乱用、滥用农药造成的农药残留问题却对我们的身体健康造成了巨大威胁。人们食用了含有农药残留的食物,严重者具有致畸性、致癌性、致突变性[2]。据调查,由于农药残留导致的癌症占比高达 60%,人体内的农残有10%通过空气、饮用水进入[3]。
现在常用的农药包括有机磷农药、有机氯农药和拟除虫菊酯农药等。其中水溶性大、吸附性能弱的农药会随水渗入地下,从而对水体和地下水体造成污染[4]。水污染又会影响到水生生物的养殖,造成鱼虾等产品的品质下降。为了保护人们的身体健康,强制性国家标准GB 3838—2002《地表水环境质量标准》对集中式生活饮用水地表水源地部分农残项目的标准限值做出了明确的规定。
通过标准查新发现,国内目前指定水中农残的测定方法主要为 GB/T 5750.9—2006《生活饮用水标准检验方法农药指标》,标准中分为填充柱气相色谱法和毛细管柱气相色谱法[5]。但单纯的气相色谱法准确性、稳定性和灵敏度都不够高,而且测定不同农残项目不但需要不同的前处理,还需要更换不同的气相色谱柱,耗费大量的时间成本和人力成本。
近些年,气相色谱–三重四级杆质谱(GC-MS/MS)技术因为能够实现农残简单快速、灵敏、准确的筛查,具有良好的重现性,能完成定性定量同步分析[6]。方法拟通过对前处理、标准溶液的配制、质谱参数等方面进行优化的方式,建立一种方便、快捷、高效的测定容易通过除草剂、杀虫剂被代入地表水中常见 32种农残 GC-MS/MS的检测方法。
1 材料与方法
1.1 材料与试剂
农残标准品:联苯菊酯、毒死蜱、氟氯氰菊酯、高效氯氟氰菊酯、嘧菌环胺、溴氰菊酯、二嗪磷、敌敌畏、苯醚甲环唑、乐果、烯唑醇、灭线磷、杀螟硫磷、甲氰菊酯、氰戊菊酯、氟氰菊酯、氟胺氰菊酯、地虫硫磷、水胺硫磷、甲基异柳磷、马拉硫磷、杀扑磷、对硫磷、甲基对硫磷、腐霉利、丙溴磷、五氯硝基苯、治螟磷、戊唑醇、三唑酮、三唑醇、三唑磷等32种农残混标(质量浓度为 50 μg/mL):曼哈格;环氧七氯 B(质量浓度为100.3 μg/mL):安谱。
实验试剂:乙酸乙酯:色谱纯,上海安谱实验科技股份有限公司;二氯甲烷:色谱纯,默克股份两合公司;石油醚、无水硫酸钠:分析纯,广州化学试剂厂。
1.2 仪器与设备
气相色谱串联质谱仪:赛默飞世尔TSQ-8000 EV;万分之一天平:赛多利斯QUINTIX124-1CN;台式冷冻离心机:赛默飞;自动浓缩仪:拜齐泰。
1.3 实验方法
1.3.1 标准溶液的配置
内标溶液:配制7 mg/L环氧七氯B内标溶液。
农药混标:配制农残混合标准的储备液,于–20 ℃保存。使用地表水的基质空白液将储备液逐级稀释为 10、50、100、200 、400和 600 μg/L标准工作溶液。
基质匹配标准工作液[7]:移取上述各浓度标准工作溶液980 μL,加入20 μL内标溶液,涡旋混匀。
1.3.2 仪器条件
1.3.2.1 色谱条件 色谱柱:TG-WAXMS气相色谱柱(30 m×0.25 mm×0.25 μm);升温程序:40 ℃保持 1.5 min,以 25 ℃/min 升至 90 ℃,保持1.5 min;以 25 ℃/min 升至 180 ℃;以 5 ℃/min升至280 ℃;以10 ℃/min升至300 ℃保持5 min;载气(He)流速1.2 mL/min,纯度≥99.999%;进样量1 µL;进样方式:不分流进样。
1.3.2.2 质谱条件 电子轰击(EI)离子源;电子能量 70 eV;传输线温度 300 ℃;离子源温度300 ℃。
1.3.3 样品处理
准确量取250.0 mL试样于500 mL分液漏斗中,分别加入30 mL二氯甲烷,剧烈振荡15 min,静止10 min分层后收集上层萃取液;再用30 mL二氯甲烷重复提取一次,将两次萃取液合并,离心吸去水分后,经无水硫酸钠干燥后分批全部转移至玻璃管,40 ℃水浴氮气吹至近干,用980 μL乙酸乙酯定容,加入20 μL环氧七氯B内标溶液[8],过0.22 μm有机相滤膜后,供GCMS/MS测定。
1.4 数据分析
数据差异显著性通过 TraceFinder软件和Xcalibur软件对检测结果进行分析。
2 结果与分析
2.1 最佳目标离子对的选择
选择合适的程序升温条件,采用Fullscan方式测定待测农残的总离子流谱图,确定其各自保留时间和母离子。选择 SRM 模式,为获得最佳质谱条件,在0~70 eV调节碰撞能量,选取丰度较高的离子碎片作为母离子。将母离子进一步裂解;选取裂解离子为子离子,优化碰撞能量[9]。由于本方法具有高选择性,可对目标化合物的母离子、产物离子、碰撞能量等质谱参数进行优化,可以筛选出多种定性离子进行确证,确保结果的准确性。32种待测农残经优化后质谱分析条件见表1,SRM模式下气相色谱串联质谱图见图1。
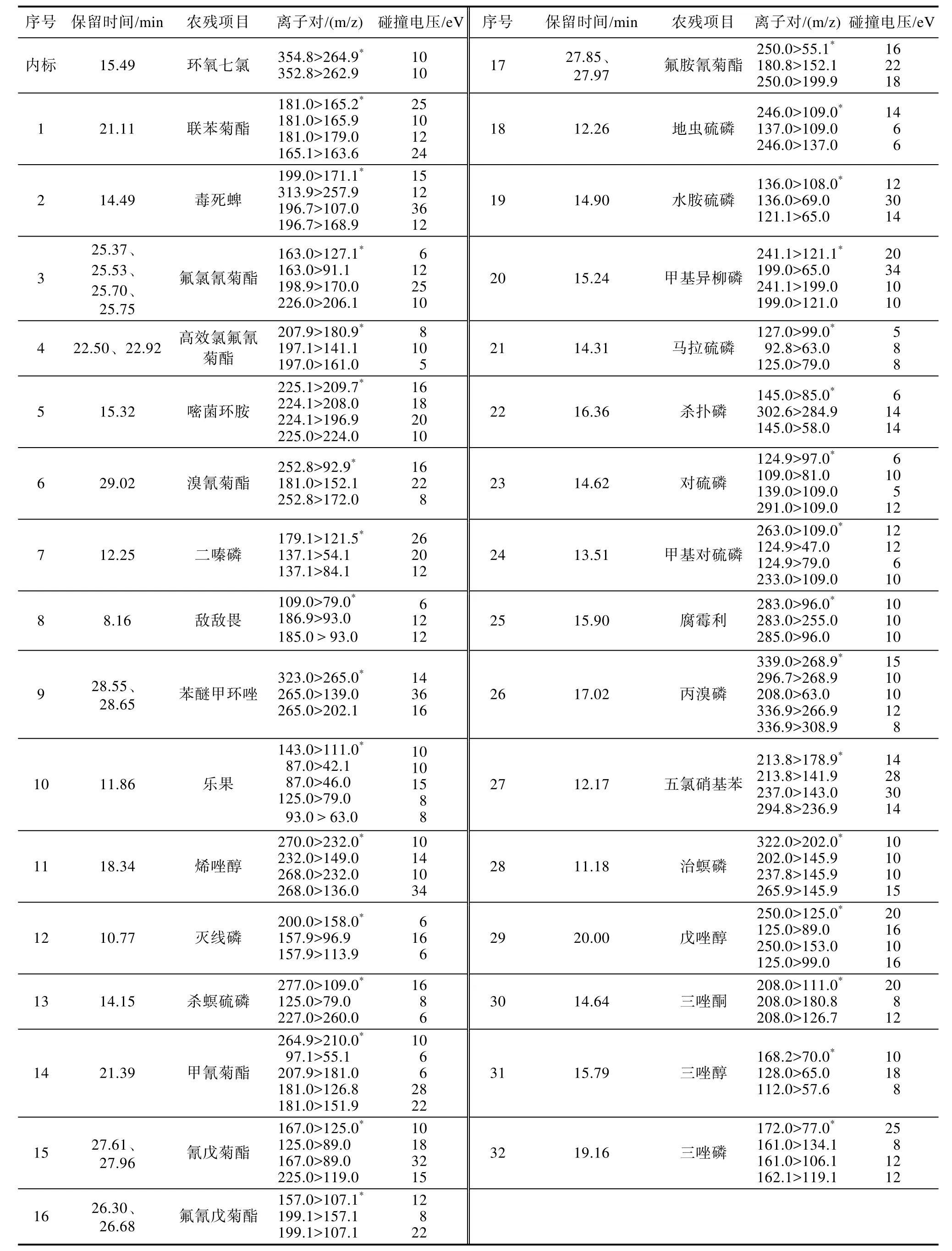
表1 32种农残和内标化合物的质谱条件Table 1 Mass spectrum conditions of 32 pesticide residues and internal standard compounds
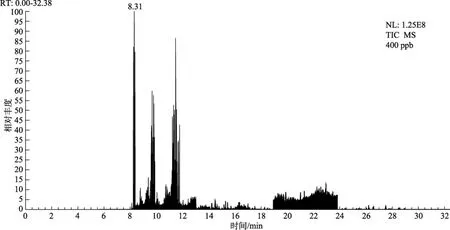
图1 400 ng/mL标准溶液谱图Fig.1 Spectrum of 400 ng/mL standard solution
2.2 前处理过程的优化
本方法通过对前处理过程中的提取试剂选择及提取次数进行优化。提取试剂选择:实验中将常见的水中农残提取液二氯甲烷与石油醚分别作为提取液进行实验,结果发现,二氯甲烷能有效的将32种农残全部提取,而在同等情况下,使用石油醚作为提取液仅能提取二氯甲烷作为提取液时敌敌畏项目的30 %左右,而乐果项目在石油醚作为提取液时完全未能有效提取,相应的乐果项目定量离子及定性离子谱图见图2和图3,因此本实验最终选择二氯甲烷作为提取液。提取次数:实验发现,当提取一次时,农残回收率很难稳定达到60%以上,提取两次或者三次时,农残回收率均能稳定达到60%,且两种提取次数的回收率相近,因此,本实验前处理采用两次提取。
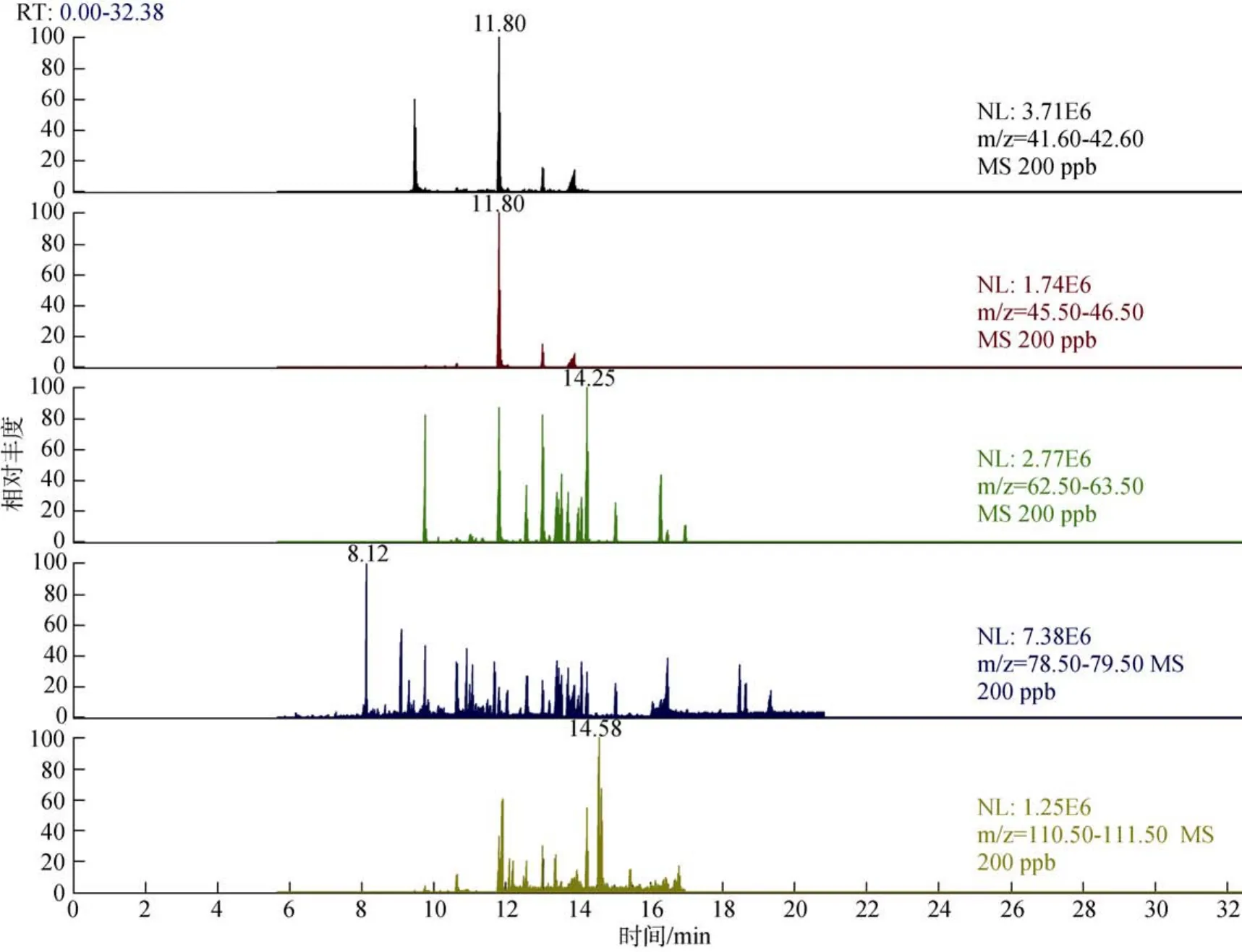
图2 二氯甲烷作为提取液时标液中乐果项目SRM谱图Fig.2 SRM spectrum of dimethoate in the time standard solution with dichloromethane as the extraction solution
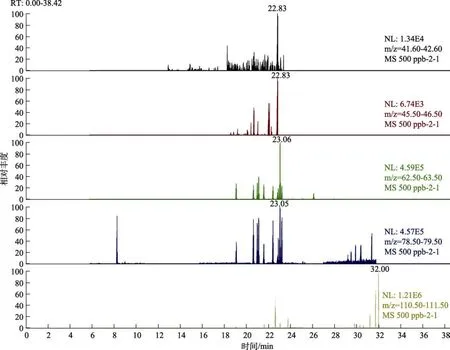
图3 石油醚作为提取液时标液中乐果项目SRM谱图Fig.3 SRM spectrum of dimethoate project in petroleum ether as extraction solution
2.3 内标的引入
在多次进平行样品的过程中,发现由于离子化效率的不稳定导致GC-MS/MS的重复性差,为了消除的离子化效率的不稳定影响,尝试在乙酸乙酯定容后加入环氧七氯B,对环氧七氯B的响应值进行折算后定量,两种方法对平行样品的响应值如下图4。通过对比可以发现,引入内标,能有效减小离子化效率的不稳定影响,避免偶然误差的发生,提高实验结果的重复性。
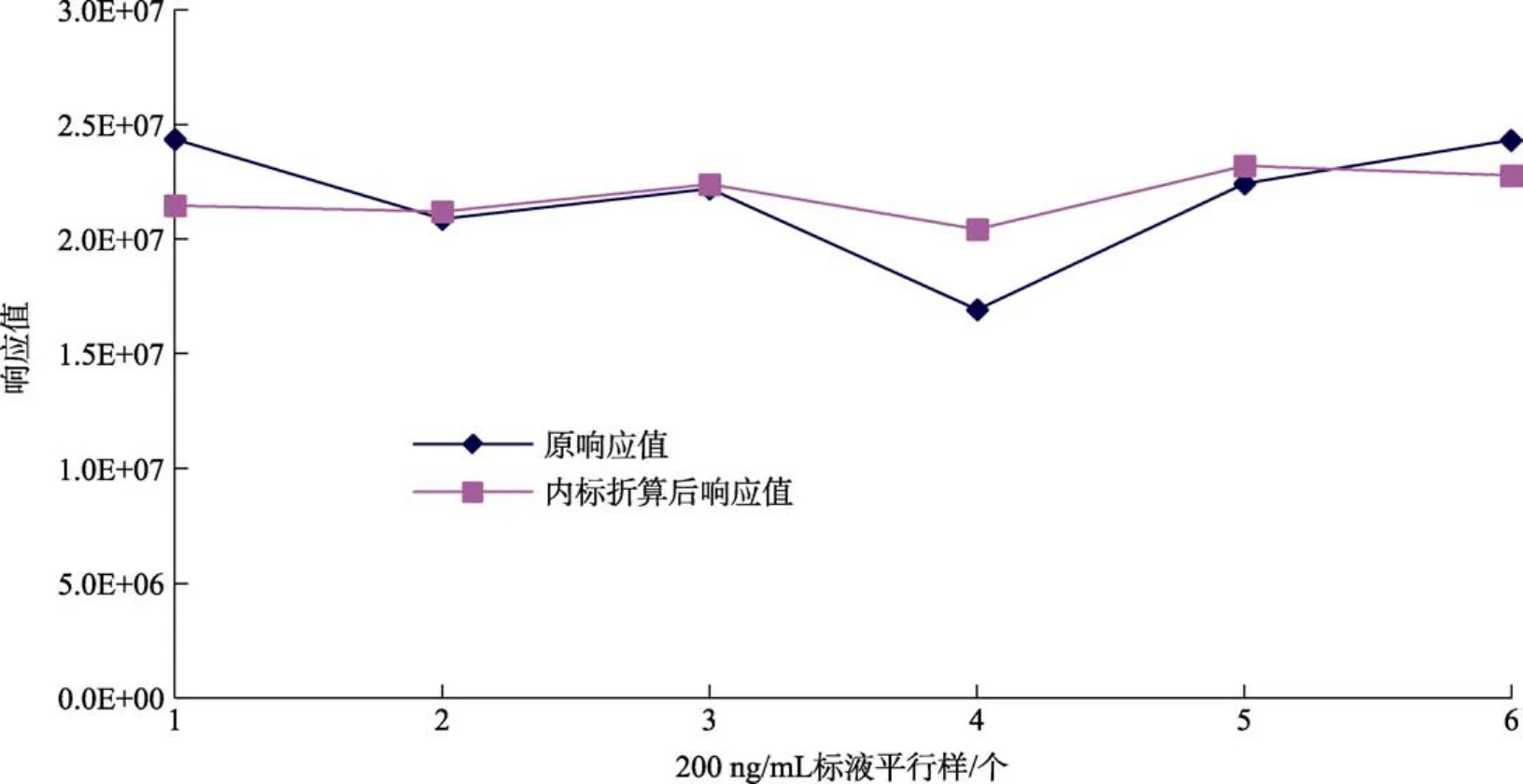
图4 200 ng/mL标液中联苯菊酯项目内标折算前后的响应值Fig.4 Response value of bifenthrin in 200 ng/mL standard solution before and after internal standard conversion
2.4 基质效应
在质谱中基质效应明显,基质效应对不同农残有不同的影响,部分农残项目在不同基质效应影响下,响应值能偏差数倍,极易导致测定结果的偏差。本实验对基质效应对目标农残检测的影响进行了探讨。通过1.5步骤,提取到地表水的基质,并配制相应的基质标液,与相同浓度下使用乙酸乙酯配制的溶剂标液做对比(结果见图5和图6),发现此实验基质标液对乐果、甲基对硫磷、杀螟硫磷、水胺硫磷、苯醚甲环唑、三唑磷有明显的增强效应,对敌敌畏、五氯硝基苯、二嗪磷、对硫磷有明显的抑制效应。为保证实验结果的准确性,配制基质曲线,进行最后的结果定量。

图5 乙酸乙酯配制的溶剂标准溶液谱图Fig.5 Spectrum of solvent standard solution prepared with ethyl acetate
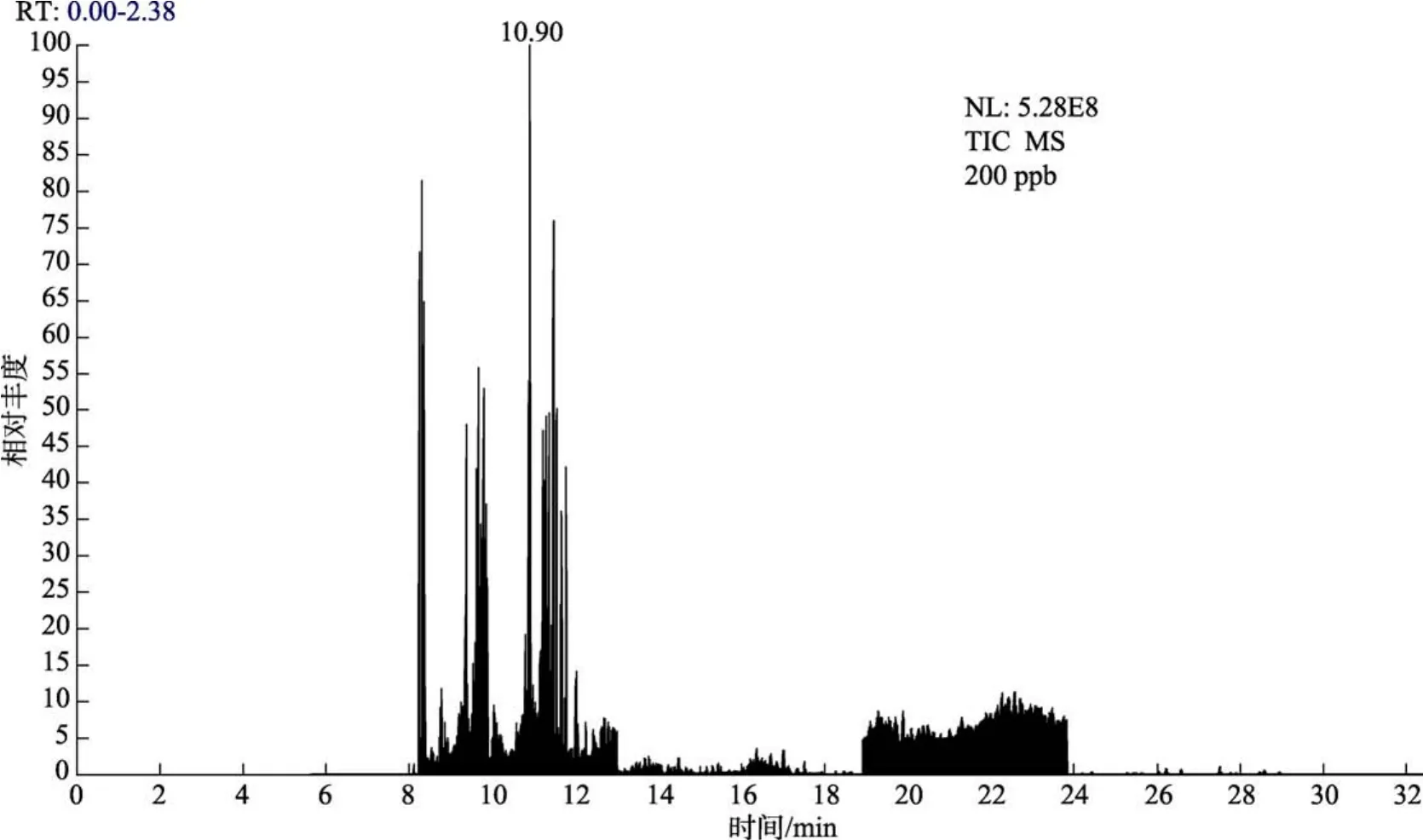
图6 地表水基质溶液配制的标准溶液谱图Fig.6 Spectrum of standard solution prepared by surface water matrix solution
2.5 线性范围
本实验采用基质匹配内标标液在相同检测条件下进行定量。在10~600 μg/L范围内,水中32种农残的线性回归方程、相关系数及最低检出限如表2所示。结果表明该检测方法的线性良好,R2≥0.995,满足GB/T 27404—2008《实验室质量控制规范 食品理化检测》要求[10]。
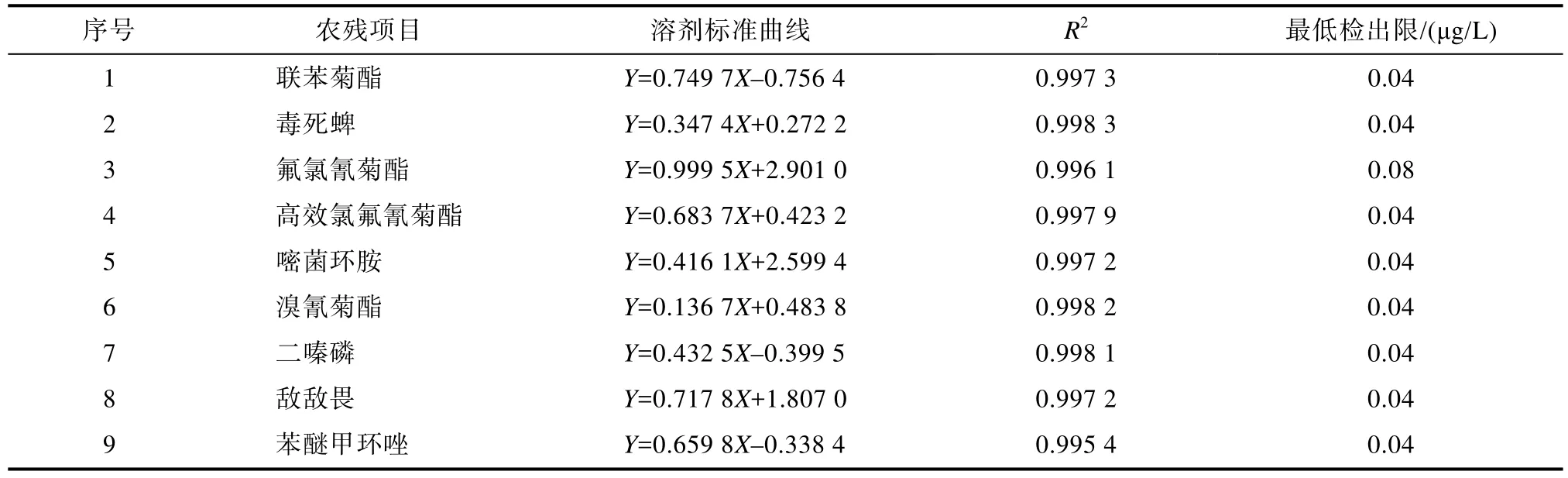
表2 水中32种农残标准曲线Table 2 Standard curves of 32 kinds of agricultural residues in water
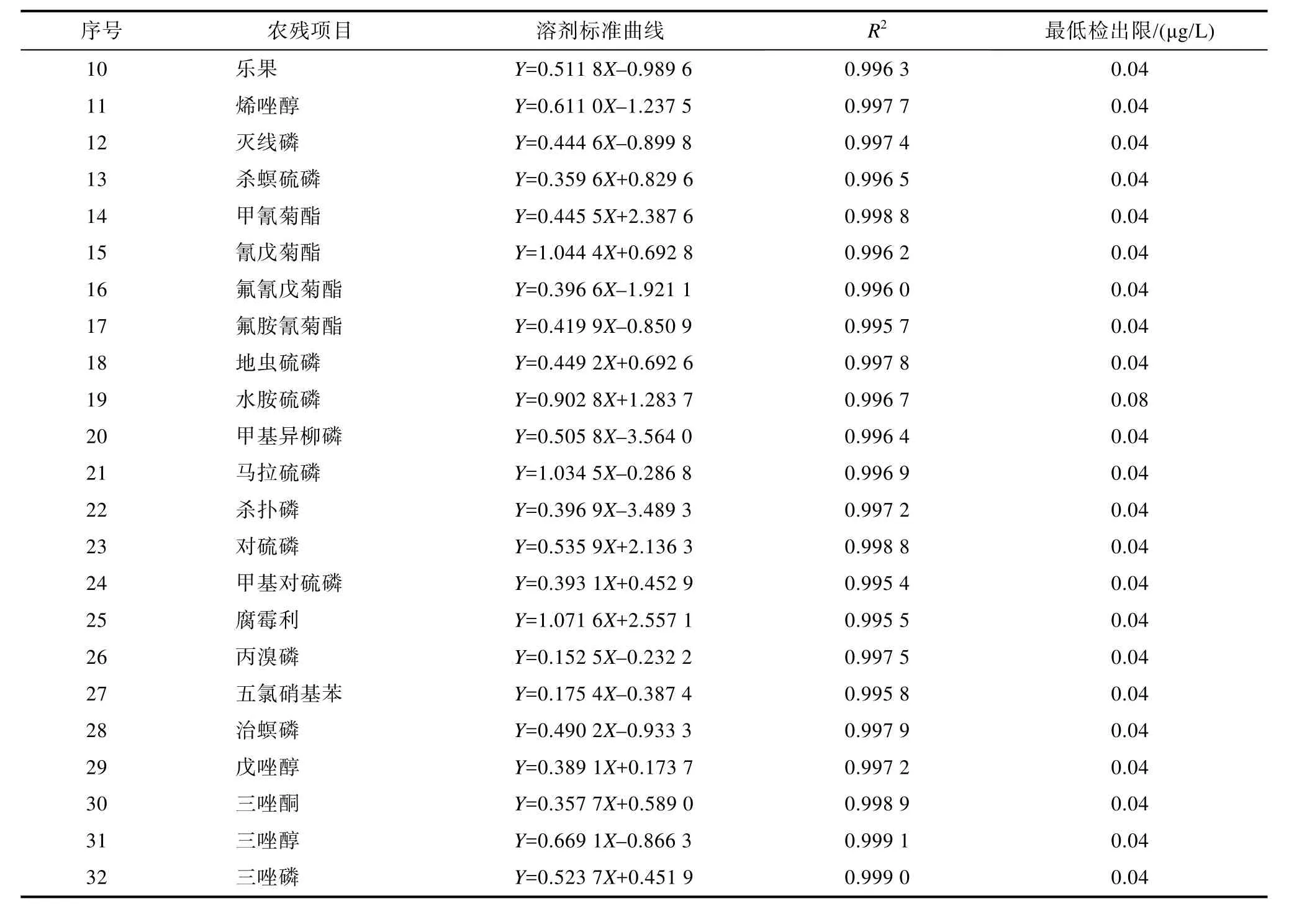
续表2
2.6 加标回收率、定量限及检出限
取空白地表水,分别添加不同浓度的混合标准溶液,获得0.2、0.4、0.8、2 μg/L添加水平样品[11],每个加标水平进行6平行实验,计算各添加水平下的回收率及相对标准偏差,见表3。在0.2 μg/L添加水平下,各农残回收率 73.9%~105.0%,相对标准偏差4.5%~17.9%;在0.4 μg/L添加水平下,各农残回收率87.5%~107.5%,相对标准偏差1.9%~12.0%;在0.8 μg/L添加水平下,各农残回收率 80.0%~108.8%,相对标准偏差3.6%~11.8%;在2.0 μg/L添加水平下,各农残回收率76.0%~106.5%,相对标准偏差1.3%~13.7%。各项农残在 0.2~2.0 μg/L添加水平下,均符合GB/T 27404—2008《实验室质量控制规范 食品理化检测》要求,因此该方法各农药定量限为0.2 μg/L。
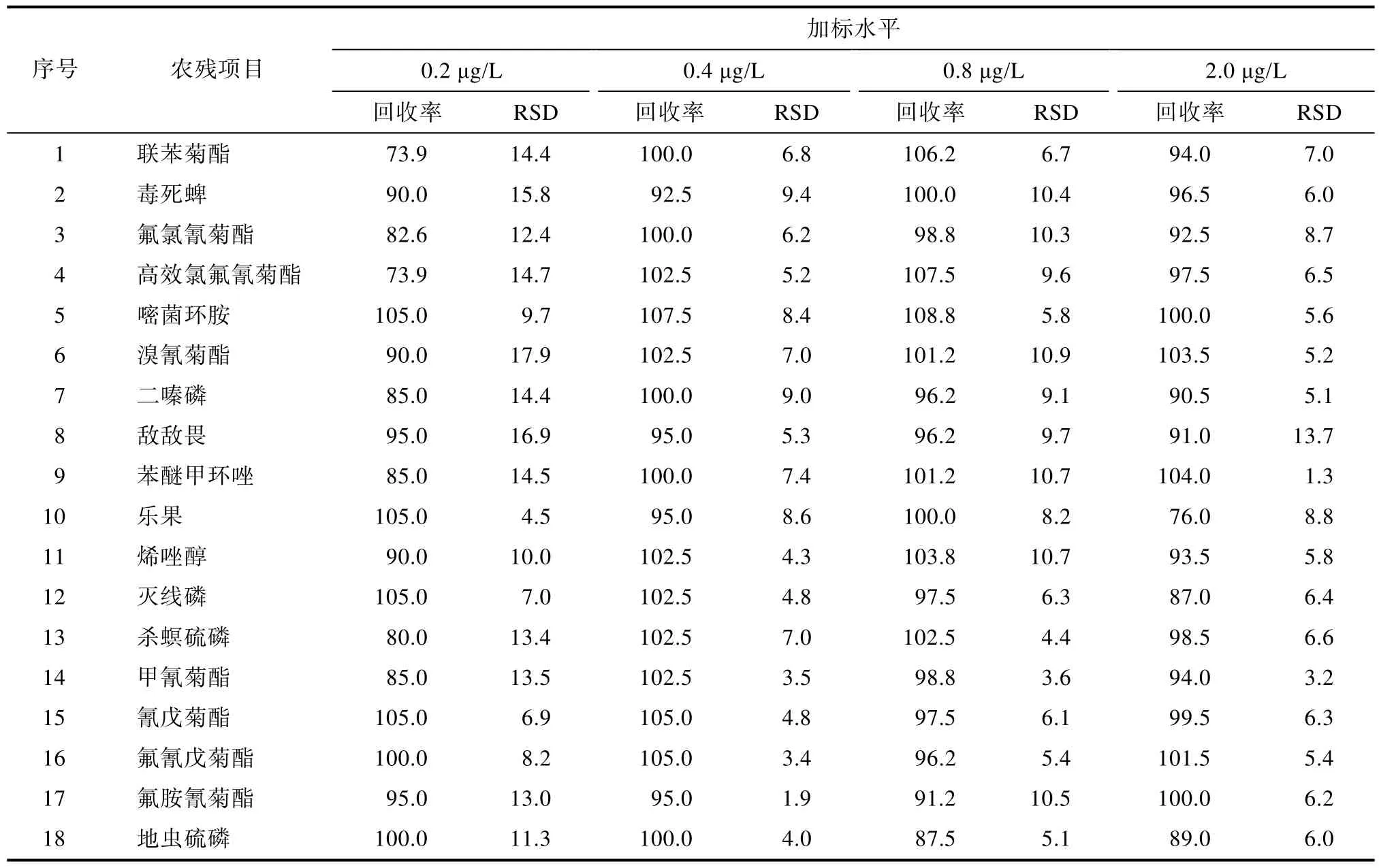
表3 加标回收测定结果Table 3 Determination results of spiked recovery %
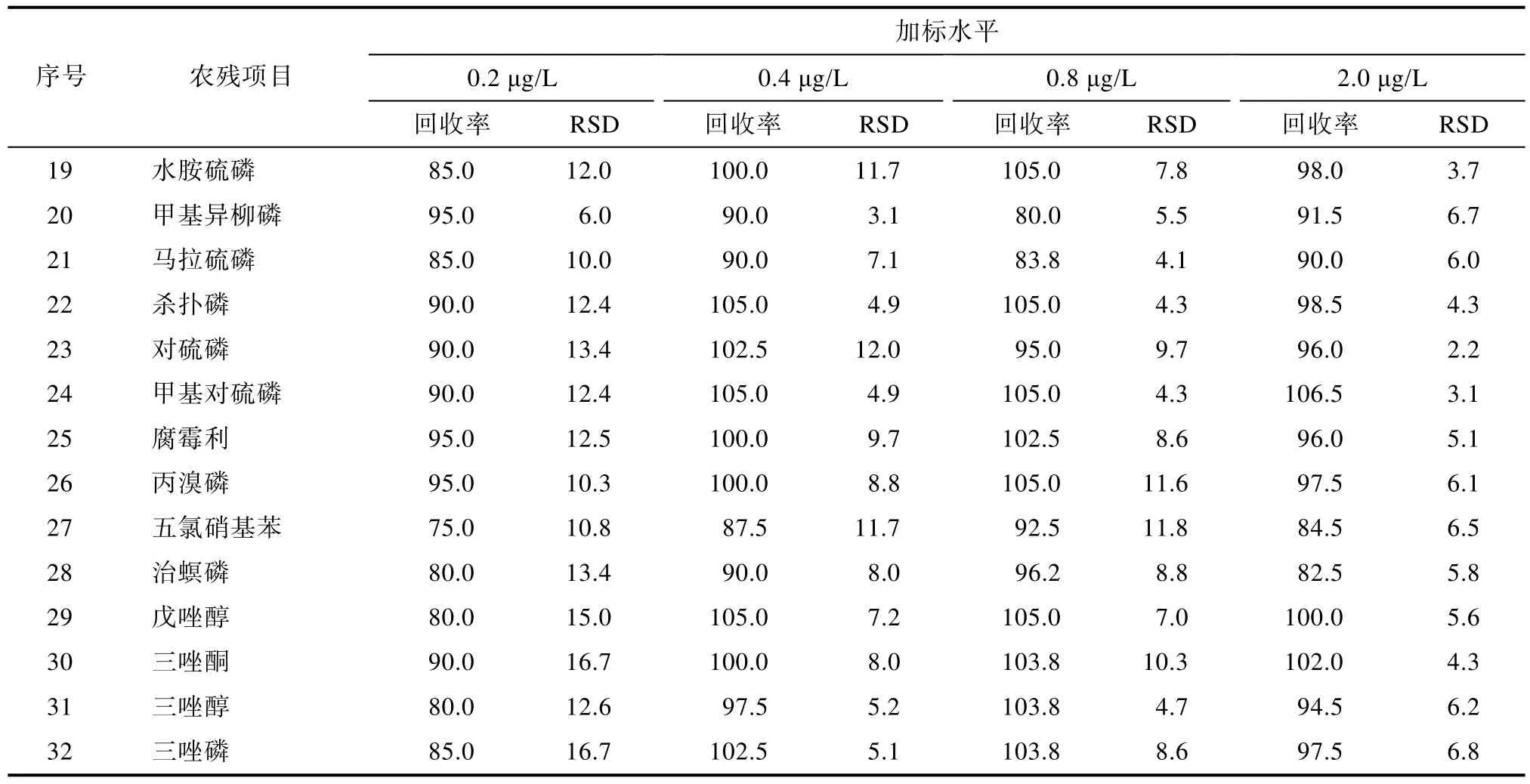
续表3
3 结论
本研究建立了同时测定地表水中 32种农残气相色谱–三重四极杆质谱(GC-MS/MS)检测方法。本方法具有高选择性,可对目标化合物的母离子、产物离子、碰撞能量等质谱参数进行优化,筛选出多种定性离子进行确证,确保结果的准确性;具有高重复性和灵敏度,一方面为避免基质效应对农残检测造成干扰,采用空白基质配制标准溶液的方式,对样品完成定量分析,另一方面为了消除的离子化效率的不稳定影响,引入环氧七氯B内标,对样品进行折算定量,因此即使方法的定量限和检出限远低于 GB/T 5750.9—2006《生活饮用水标准检验方法农药指标》和 GB 3838—2002《地表水环境质量标准》要求的情况下,准确度和精密度均能满足标准要求。同时,该方法前处理简单,使用一种提取方式,能一次性提取32种农残进行检测,节约大量人力和时间,适用于地表水中多种农药残留的日常定量分析。
猜你喜欢
化学工程师(2022年3期)2022-04-19
口腔护理用品工业(2021年4期)2021-11-02
上海化工(2021年2期)2021-04-23
中国粮油学报(2019年4期)2019-07-12
中成药(2018年6期)2018-07-11
中成药(2018年2期)2018-05-09
中成药(2017年4期)2017-05-17
中国粮油学报(2016年5期)2016-01-23
科技与企业(2015年20期)2015-10-21
西南军医(2015年1期)2015-01-22
