
核桃品种及近缘树种鉴定的DNA条形码开发
2022-11-24 06:46:36张焕玲
森林工程 2022年6期
张焕玲
(西北农林科技大学 林学院,陕西 杨凌 712100)
0 引言
核桃(Juglansregia)属胡桃科(Juglandaceae)核桃属(Juglans)落叶乔木树种,是我国主要的木本油料和干果树种,居世界4大坚果之首。20世纪50年代我国开始核桃引种和选育工作,目前核桃栽培面积和年产量均居世界首位,栽培的核桃品种已达500多个[1]。但核桃栽培中品种管理混乱,以次充好、种苗混杂现象严重,不但影响核桃栽培的经济效益,而且损害了良种持有人和种植户的利益[1]。因此,快速、准确地开展核桃品种鉴定,对促进核桃产业健康发展具有重要意义。目前,核桃品种鉴定主要依据形态特征,结合物候特点及同工酶等指标来进行[2-4]。由于核桃品种间本身形态差异小,且种苗交易多在非果期,仅通过枝、叶和树皮等表型特征很难准确鉴别品种,急需新的快速鉴定技术。
DNA条形码技术(DNA barcoding)是指可以利用生物体基因组中的一个或者几个能够代表该物种的、有足够变异的、易扩增且相对较短的DNA片段,快速实现物种鉴别的一种方法[4-6]。该技术具有操作简单、准确性高和稳定性好等特点,目前已广泛应用于动植物和微生物的鉴定中,并有力地推动了物种分类和系统发育学科的快速发展[6-14]。
高等植物DNA条形码研究主要集中在叶绿体基因组和核基因组,叶绿体基因DNA片段如matK、rbcL、psbA-trnH以及核糖体DNA片段ITS是目前广泛应用的DNA条形码[15-19]。在胡桃科的相关研究中,陈亚辉等[20]应用DNA条形码对不同品种美国山核桃进行研究,发现ITS和matK序列可以有效鉴别不同品种。宋艳波等[21]基于matK基因序列对核桃的早、晚食品种进行鉴别。朱凤琴等[9]发现利用ITS序列可区分核桃、核桃楸、山核桃以及枫杨属的相关物种。本研究利用国际生物条形码联盟(CBOL)推荐的4条首选DNA条形码序列,对生产中广泛栽培的11个核桃品种和5个近缘树种开展分子鉴定,以期筛选稳定性好、适应性高的DNA条形码,为核桃品种的快速鉴定和核桃产业的健康发展提供技术支持。
1 材料与方法
1.1 实验材料
实验材料包括14份核桃属树种(11个核桃品种和3个核桃种)、1个枫杨属树种和1个山核桃属树种,见表1。均来自西北农林科技大学渭河试验站核桃种质资源圃。

表1 实验材料编号及信息Tab.1 Material number and information
每树种(品种)分别采集5个样株,样株为当年生新鲜健康叶片100 g(作为5次生物学重复),置于装有硅胶颗粒的自封袋内,带回实验室于-80 ℃超低温冰箱保存备用。
1.2 总DNA的提取
用改良的十六烷基三甲基溴化铵法(cetyl trimethyl ammonium bromide,CTAB)提取总DNA:在2 mL离心管中加入CTAB提取缓冲液1 mL,65 ℃水浴预热;取叶片0.2 g,加入液氮快速研磨;在预热的CTAB提取缓冲液中加入20 μLβ-巯基乙醇,倒入叶片研磨粉充分混匀,后置于65 ℃水浴中保温45 min,其间轻轻晃动离心管数次;离心管中加入等体积氯仿和异戊醇(其比例为24∶1),轻轻颠倒混匀,4 ℃下12 000 r/min离心10 min,移上清至另一个新的2 mL离心管中(此步骤重复2次);离心管中加入2倍体积的无水乙醇于-20 ℃放置30 min后,12 000 r/min离心10 min回收DNA沉淀;弃上清,用70%乙醇清洗沉淀2次,室温干燥DNA沉淀;将DNA溶于100 μL的TE缓冲液中溶解,加入RNA水解酶10 μL去除RNA,4 ℃保存备用。
1.3 目的片段的扩增、回收与测序
本研究选用国际生物条形码联盟(CBOL)推荐的3个叶绿体基因组候选条形码rbcL、matK和trnHpsbA片段,以及1个核基因组候选条形码ITS1。4个候选条形码的通用引物从美国国家生物技术信息中心(National Center for Biotechnology Information, NCBI)查询,由上海生工合成,引物信息及相应PCR反应条件详见表2。

表2 4个候选条形码的引物序列及扩增条件Tab.2 Primer sequences and amplification conditions of four candidate barcodes
PCR反应总体积60 μL:其中1对引物各3 μL,Taq Master Mix 30 μL,模板DNA 6 μL,超纯水18 μL。 扩增产物经0.1 %的琼脂糖凝胶100 V电压下电泳分离,凝胶成像系统观察记录电泳结果。将凝胶上的目的条带切下后用Monarch DNA回收试剂盒回收目的片段,送西安擎科泽西生物科技责任有限公司进行双向测序,测序引物同PCR引物。
1.4 序列校对与分析
测序后的序列经DNAman10.0软件拼接、校对。然后用MEGA-X软件对测序文件进行手动调整,最后测序结果以FASTA格式输出。用MEGA-X分析序列碱基组成、GC 含量、变异位点、简约信息位点。
1.5 遗传距离估算与系统发育分析
以变异位点最丰富的条形码序列为数据,将处理与分组后的数据导入MEGA-X软件,进行Clustal W对比,采用Kimura2-parameter(K2-P)模型计算遗传距离,以最大简约法(Maximum Parsimony,MP)构建系统发育树,用1 000次重复bootstrap检验各分支的支持率,自展支持率需大于等于50%。
2 结果与分析
2.1 4种候选条形码的PCR扩增与测序结果
4种候选条形码在16个 树种(品种)中均获得了预期的扩增条带,扩增成功率为100%,如图1所示。
测序结果表明,rbcL序列在样品间差异极小,仅枫杨与其他树种存在一个碱基的差异(597位枫杨为A,其他树种为G),11个核桃品种之间的rbcL序列完全一致,故在后续实验分析中排除rbcL序列。psbA-trnH序列只在属间存在差异,在属内序列一致性为100%,不符合作为DNA条形码进行品种鉴定的要求,故排除。
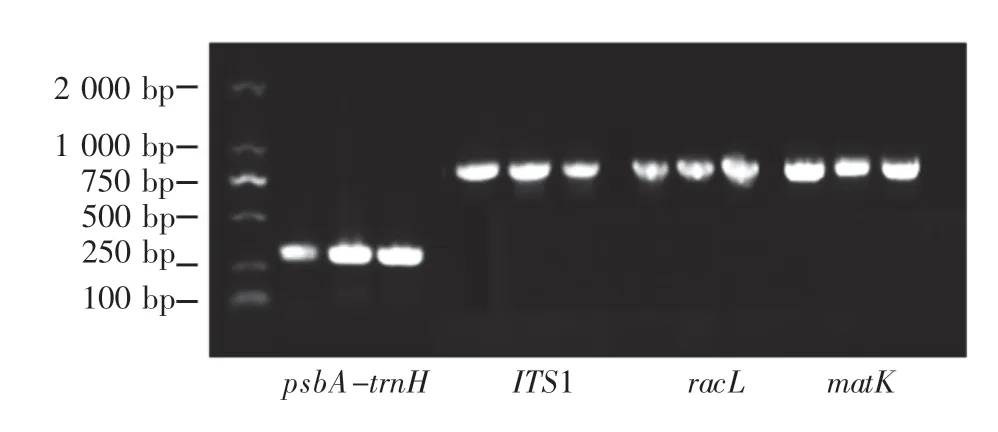
图1 4条候选条形码的部分扩增结果Fig.1 Partial amplification results of four candidate barcodes
ITS1和matK序列在树种(品种)内序列一致性为100%,在树种(品种)间序列有差异。不同树种的ITS1和matK序列长度以及碱基G、C含量见表3,大小分别为696~701 bp和612~619 bp。2条序列的碱基G、C含量差异较大,ITS1序列碱基G、C含量为56.02%~58.71%,而matK序列碱基G、C含量为34.05%~34.74%,ITS1序列的G、C含量明显高于matK序列,说明matK序列发生变异的可能性要比ITS1序列大,ITS1序列则相对稳定。
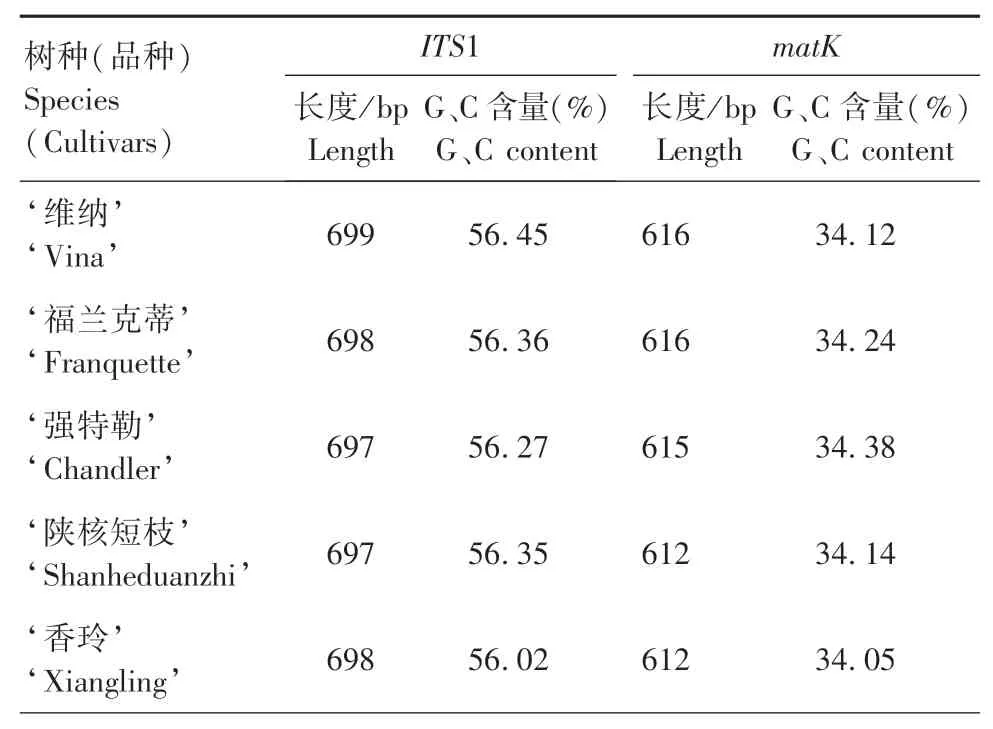
表3 不同树种(品种)ITS1和matK条形码序列的长度及GC碱基含量Tab.3 Sequence length and GC content ofITS1 and matK in different species(cultivars)

续表3
2.2 ITS1和matK序列变异分析
应用MEGA-X对各序列进行对比,发现16个树种(品种)间在matK序列中共发生18个碱基位点变异,见表4,其中单核苷酸变异位点3个,简约性信息位点数15个,有2处发生碱基缺失,序列一致性为99.12%。matK序列在属间的差异明显大于种间或品种间,枫杨和碧根果中的变异位点要明显多于其他核桃品种或核桃属树种。在11个核桃品种中,除‘维纳’和‘六盘水’品种matK序列一致外,其他9个核桃品种matK序列均存在差异。
16个树种(品种)的ITS1序列变异位点相比matK序列要少很多,只有8个碱基位点变异,见表5,其中单核苷酸变异位点2个,简约性信息位点数6个,序列一致性为99.88%。除了‘维纳’与‘清香’和野核桃的ITS1序列完全一致外,其他9个核桃品种及4个近缘树种间的ITS序列均存在差异,matK不能区分的‘维纳’和‘六盘水’品种可以被ITS1区分。
综上,matK结合ITS1序列可以鉴定所有供试的核桃品种及近缘种,是核桃品种及近缘种鉴定的理想DNA条形码。
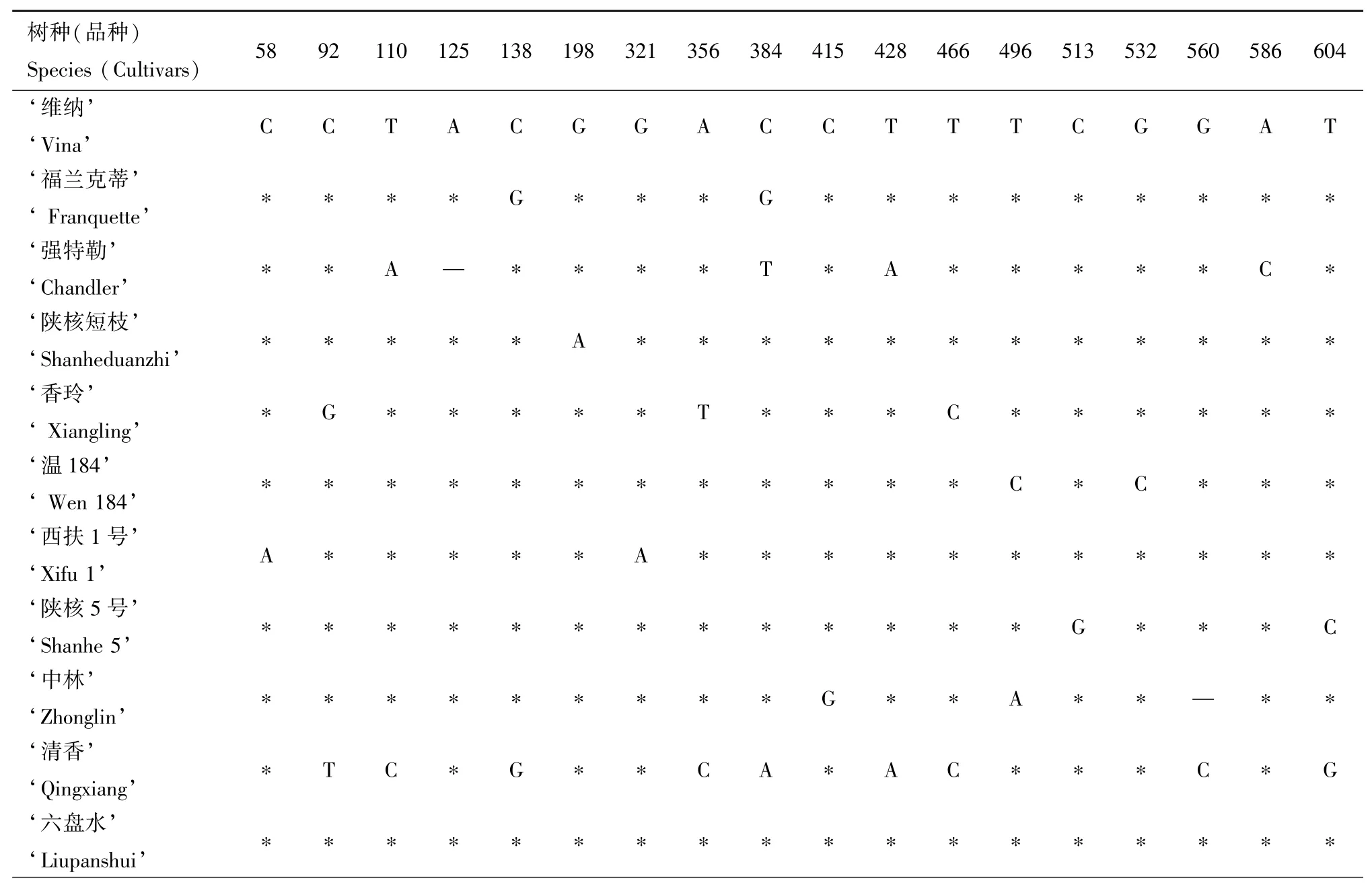
表4 不同树种(品种)的 matK序列变异位点Tab.4 The variable sites of matK sequence of different species(cultivars) bp
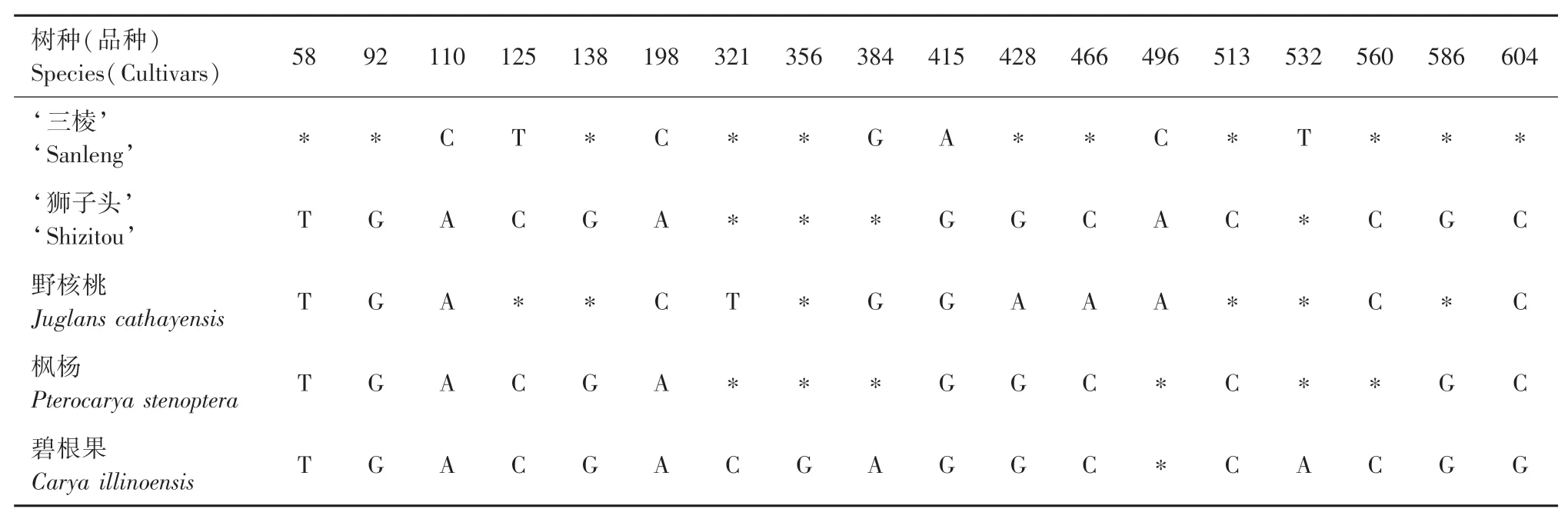
续表4

表5 不同树种(品种)的ITS1序列变异位点Tab.5 The variable sites of ITS1 sequence of different species (cultivars)bp
2.3 遗传距离及系统发育分析
为了验证筛选条形码的鉴定准确性,本文基于matK序列,以Kimura-2参数模型计算了不同树种(品种)间的遗传距离见表6,并以最大简约法(Maximum Parsimony,MP)构建了16个树种(品种)的系统发育树,如图2所示。
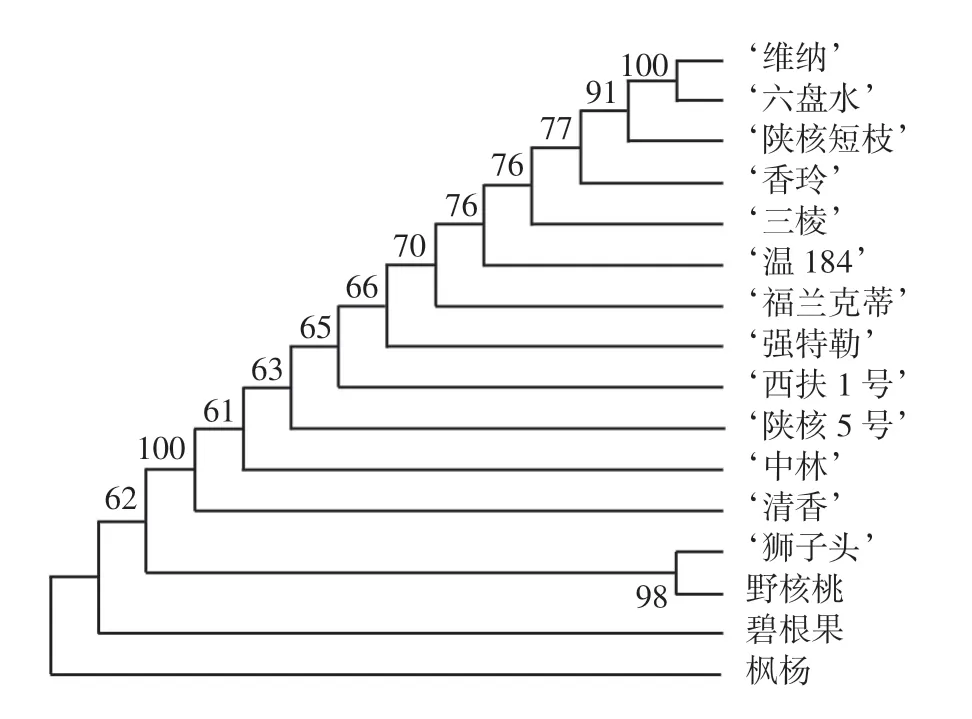
图2 基于matK序列构建的MP系统发育树Fig.2 Maximum parsimony tree based on matK barcode
结果表明,核桃品种间的遗传距离要明显小于属间和种间的遗传距离。11个核桃品种的遗传距离为0.000 0~0.009 7,‘六盘水’与‘清香’、‘清香’与‘维纳’的遗传距离最大,均为0.009 7,而‘六盘水’与‘维纳’的遗传距离为0。8个国内核桃品种中,‘中林’与‘香玲’的遗传距离最大,为0.008 3,‘陕核5号’和‘温184’遗传距离最小,为0.001 1。与近缘种相比,核桃品种与碧根果的遗传距离最大,与‘狮子头’之间的距离次之,与野核桃和枫杨之间的遗传距离相对最小。
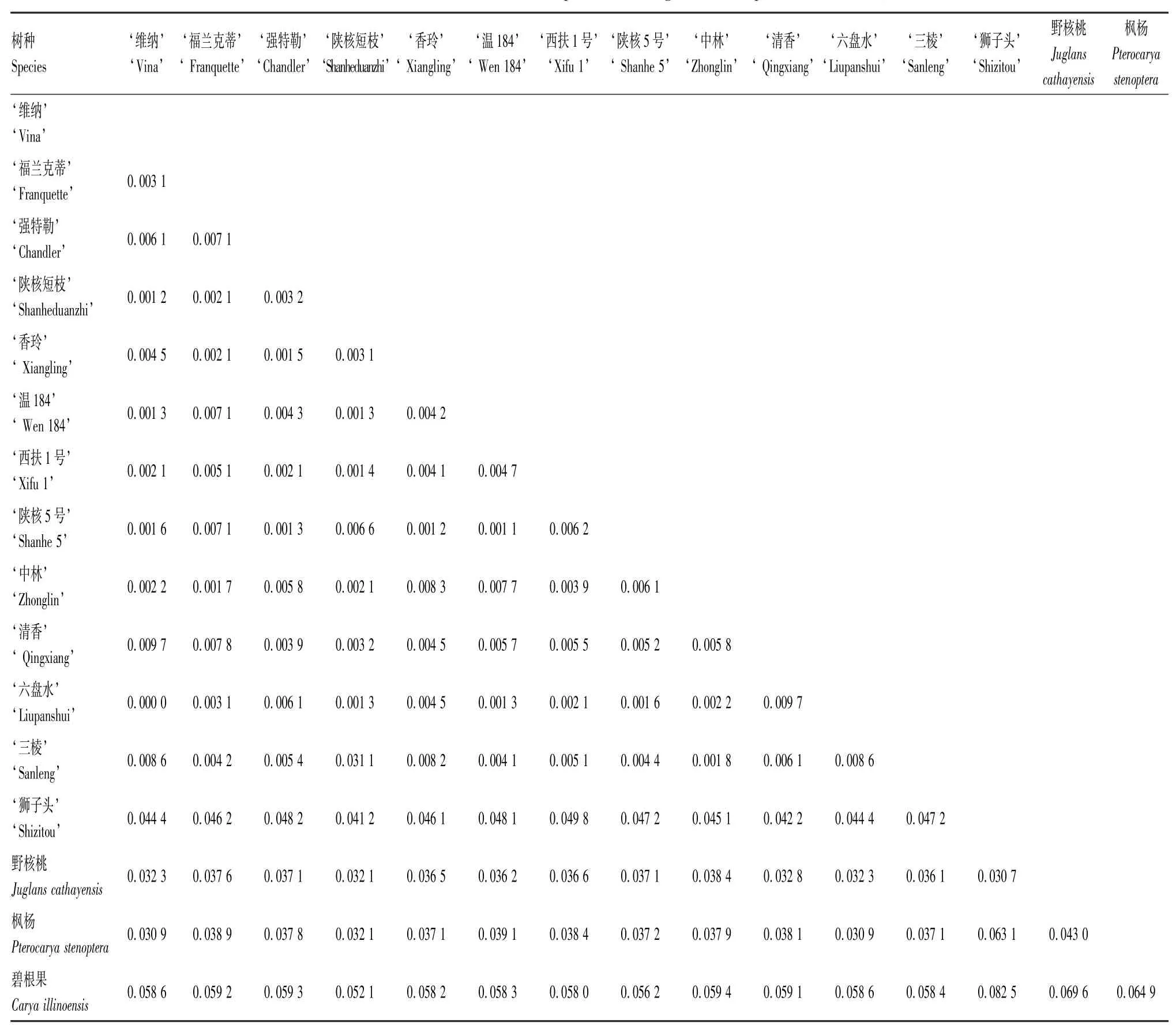
表6 不同树种(品种)matK序列之间的遗传距离Tab.6 Genetic distances of matK sequences among different species (cultivars)
MP系统发育树显示,除‘维纳’和‘六盘水’外,其他不同核桃品种都能明显区分,表明matK条形码可准确鉴定82%的核桃品种,而且基于matK条形码构建的系统发育树能准确区分胡桃科枫杨属、核桃属和山核桃属的遗传进化关系,可为胡桃科植物区分或育种研究提供依据。
3 结论与讨论
本研究以国际生物条形码联盟(CBOL)推荐的4个DNA序列为候选条形码,通过扩增、测序和序列分析,筛选确定了matK+ITS1条形码组合可有效鉴别11个核桃品种及核桃属与枫杨属、山核桃属树种,弥补了胡桃科树种传统形态学分类的不足,对研究核桃属植物的系统分类、群体演化和资源开发,确保核桃产业良种化健康发展具有重要作用。
DNA条形码技术因其操纵简单、结果稳定,在物种和品种鉴定中被广泛采用。尽管CBOL推荐了不少首选DNA条形码,但同一条形码在不同树种中的扩增成功率、不同条形码的引物通用性、条形码的变异性大小均存在差异[19],实际应用中必须开发适合目的树种的最佳条形码。
rbcL是叶绿体基因组中编码核酮糖-1,5-二磷酸羧化/加氧酶大亚基的基因,由于有容易扩增、通用性好、易于比对序列等特征,故被提议为候选的DNA条形码序列[7],但该基因变异性很小,很难区别种级水平的物种[18],本研究也证实了这一点,除了枫杨有一个位点变异外,其他15个树种rbcL序列完全一致。psbA-trnH是叶绿体基因组中进化速率最快的基因间隔区之一,其psbA端的序列为编码基因,有较高的保守性,便于设计广泛应用的引物,且容易扩增[8]。但由于该片段中有较多的polyA/T结构,并且插入/缺失现象很常见,引起不同物种间的序列长度相差较大,造成使用psbA-trnH片段进行对非同属类群间比较时有困难[19],本文结果也表明psbA-trnH序列只在属间有差异,在属内完全一致。
matK基因是叶绿体基因组编码蛋白质基因中进化较快的基因之一,其扩增效率一直是作为通用条形码序列的争议问题[16],但该基因在美国山核桃品种鉴定中扩增效率和引物通用性都较好[19],也是本研究中鉴定核桃品种及其近缘树种效果最理想的条形码。ITS1序列是位于18S与5.8S rRNA编码基因之间的片段,具有拷贝数高、易扩增的特点,是美国山核桃品种鉴定中变异位点最多的条形码[19]。本研究中,核桃品种及其近缘种的ITS1序列变异没有matK序列丰富,但其可以作为补充条形码鉴别matK序列不能鉴别的核桃品种。
核桃是我国栽培面积最广的经济林干果之一,核桃品种之间的形态特征差异小,分子手段鉴定品种十分必要。本文只从4个候选条形码序列中开展了11个核桃品种和5个近缘种的DNA条形码筛选研究,扩大候选条形码和树种(品种)范围,筛选更多的DNA条形码并验证筛选的2种条形码的通用性是下一步研究的重点。
猜你喜欢
今日农业(2021年19期)2021-11-27 00:45:49
少年文艺·开心阅读作文(2021年8期)2021-09-05 02:57:46
小学科学(学生版)(2019年5期)2019-05-21 01:00:22
少儿美术(快乐历史地理)(2019年11期)2019-04-20 12:33:20
基层中医药(2018年2期)2018-05-31 08:45:17
现代园艺(2018年2期)2018-03-15 08:00:35
小学生导刊(2017年13期)2017-06-15 20:29:38
中国林业产业(2016年5期)2016-04-03 00:33:02
中国林业产业(2016年5期)2016-04-03 00:32:38
武夷学院学报(2014年5期)2014-07-19 10:08:27
