
水系锌二次电池MnO2正极的晶体结构、反应机理及其改性策略
2022-11-24 11:16陈鲜红阮鹏超吴贤文梁叔全周江
物理化学学报 2022年11期
陈鲜红,阮鹏超,吴贤文,梁叔全,2,周江,2,*
1中南大学材料科学与工程学院,长沙 410083
2中南大学电子封装及先进功能材料湖南省重点实验室,长沙 410083
3吉首大学化学化工学院,湖南 吉首 416000
1 引言
当前,能源危机和环境污染等问题日益突出,开发可持续发展的新能源成为当务之急。而太阳能、潮汐能、风能及生物能源等可再生能源易受环境影响,具有间歇性和不可控性。电化学储能技术作为新一代绿色能源技术,无需使用化石燃料,可有效减少温室气体的排放,是目前全球科研界的聚焦点之一。其中水系二次电池由于成本低、安全性高、毒性小,环境友好等优势受到广泛关注1。相较于众多金属如钠2、钙3、锂4,5等,金属锌在水系电解液中具有更高的可逆性和稳定性6,同时,锌还具有丰度高、理论容量高(820 mAh·g−1)及氧化还原电位低(−0.763 V vs.标准氢电极)等优势,在规模储能领域中展现出广阔的发展前景7,8。
水系锌二次电池的电化学性能与正极材料的结构和形貌特征息息相关9,10,开发容量高、电化学稳定的正极材料是发展高性能锌二次电池储能器件的关键因素之一。现已报道的正极材料有锰基氧化物11,12、钒基氧化物13,14、普鲁士蓝类似物15、有机化合物16、层状硫化物17、聚阴离子化合物18及chevrel相化合物19,20等(图1a)。其中MnO2具有种类丰富、毒性小、成本低、容量高(616 mAh·g−1,基于2个电子转移数)、放电平台高(大于1.35 V vs.Zn2+/Zn)等诸多优势,被作为正极材料广泛地应用于水系锌二次电池中21。然而,目前报道的水系锌锰电池仍无法达到预期的要求,其不令人满意的电化学性能以及多样的储能机制在很大程度上取决于MnO2的晶体结构(α、β、γ、δ、λ和R态等,图1b)22,23和电解液条件24,25。
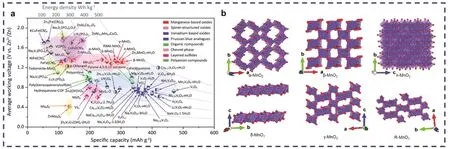
图1 (a)锌电池具有代表性的正极材料的工作电压和比容量20;(b) α-MnO2,β-MnO2,γ-MnO2,λ-MnO2,δ-MnO2和R-MnO2的晶体结构示意图23Fig. 1 (a) The working voltage and specific capacity of representative cathode materials for zinc battery 20.(b) Schematic diagram of crystal structures of α-MnO2, β-MnO2, γ-MnO2, λ-MnO2, δ-MnO2 and R-MnO2 23.
本文主要总结了MnO2的晶体结构和储能机制,以及针对MnO2主要的两类储能机制(嵌入-脱出和溶解-沉积机制)工作时所存在的电池容量低、循环寿命短等问题所提出的优化策略,为高性能锌锰二次电池的发展提供参考。
2 二氧化锰的晶体结构
二氧化锰具有多种晶体结构,如α-MnO2,β-MnO2,γ-MnO2,λ-MnO2,δ-MnO2和R-MnO2。在这些晶体结构中,每个Mn4+离子被6个相邻的氧离子包围,形成基本八面体单元MnO6,这些单元通过周期性共享顶点/边缘而形成不同的晶体结构类型(链/隧道/层状),被作为正极材料广泛应用于水系锌二次电池中22。本节将详细介绍不同类型MnO2的晶体结构特征(表1)。
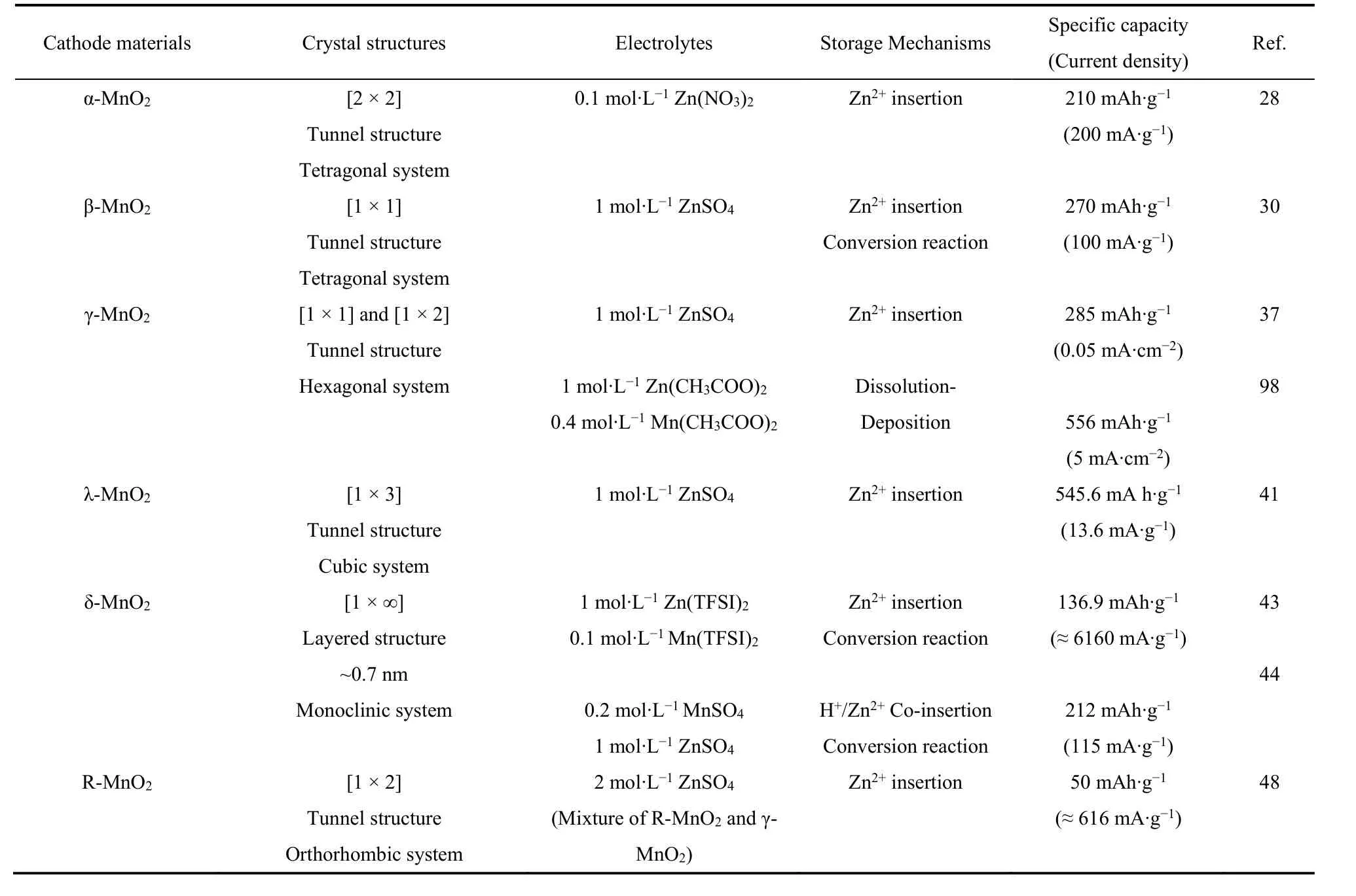
表1 不同晶型MnO2的晶体结构、储能机制及其性能Table 1 The crystal structure, energy storage mechanism and performance of different crystalline MnO2.
2.1 α-MnO2
α-MnO2具有[2 × 2,~0.46 nm × 0.46 nm]隧道结构,属于体心四方晶系,空间群为I4/m,其基本八面体单元MnO6通过共享角形成双链结构。α-MnO2较宽的隧道结构可允许Zn2+沿z轴快速嵌入/脱嵌26。纳米尺寸的α-MnO2直径小,具有较大的比表面积和更高的电解液可及性,能有效地提高电池的容量27。2009年Xu等28报道了α-MnO2在弱酸性Zn(NO3)2电解液中Zn2+的嵌入/脱嵌行为,之后又证明了放电时Zn2+会嵌入α-MnO2正极中形成ZnMn2O4,充电过程为其逆反应,从而实现了210 mAh·g−1的高容量(电流密度为200 mA·g−1),表明在α-MnO2中Zn2+嵌入-脱嵌行为的高度可逆性29。
2.2 β-MnO2
β-MnO2具有[1 × 1,~0.23 nm × 0.23 nm]隧道结构,属于正方晶系,为金红石结构22,其晶胞中的氧原子呈紧密排列的六角形结构,其中锰原子占据八面体的中心,氧原子分别占据八面体的六个顶点。β-MnO2的隧道结构较窄,其热力学稳定,理论上不利于Zn2+的嵌入。但Islam等30设计了一种暴露(101)面的β-MnO2纳米棒,这种独特的棒状形态有助于Zn2+嵌入/脱嵌。利用原位X射线衍射技术(in situ XRD)可观察到β-MnO2在首次经历Zn2+嵌入时,(101)晶面的峰值会略微向较低的角度移动,并在完全充电后恢复,不存在明显的结构畸变。这种改性的电极材料在100 mA·g−1电流密度下实现了270 mAh·g−1的高比容量。同时,近年来,研究人员利用缺陷工程、复合材料等策略,通过改变材料晶格结构或调节活性位点等方式,显著提高了β-MnO2的电化学性能31,32。
2.3 γ-MnO2
γ-MnO2由交替生长的[1 × 1,~0.23 nm × 0.23 nm,铁矿岩]和[1 × 2,~0.23 nm × 0.46 nm,菱岩]隧道组成,属于六方晶系33。γ-MnO2隧道结构的无序性导致其具有较低的结晶度,同时存在较多缺陷,包括孪平面(Tw或Mt)、Mn(III)取代Mn(IV)和Mn空位等34。1986年γ-MnO2首次被作为正极应用于弱酸性水系锌二次电池中35,之后Kumar等36将一种聚(偏氟乙烯)-三氟酸锌凝胶聚合物作为电解液,尝试进一步分析Zn2+在γ-MnO2的嵌入/脱嵌行为,遗憾的是其深入机理未能明晰。直到2015年,Alfaruqi等37研究表明,Zn2+嵌入γ-MnO2时会发生相变,形成层状L-ZnyMnO2、ZnMn2O4和γ-ZnxMnO4,放电时又重新恢复成γ-MnO2,阐明了充放电循环时γ-MnO2的具体储能过程。此外,有报道指出,可通过调节温度来控制γ-MnO2向γ/β-MnO2发生结构转变,而γ/β-MnO2具有比γ-MnO2更优异的电化学性能,这可为设计高性能二氧化锰材料提供参考38。
2.4 λ-MnO2
λ-MnO2具有[1 × 3]隧道结构,是一种典型的尖晶石结构,Mn和O离子分别占据八面体的16d位和32e位39。1981年Hunter成功制备λ-MnO2,即先采用前驱体法制备锰酸锂,再利用酸性溶液处理,使其发生氧化脱锂反应,最后生成λ-MnO240。λ-MnO2隧道结构尺寸有限,在一定程度上会阻碍Zn2+的快速嵌入/脱嵌,但Yuan等41将由稀酸溶液处理LiMn2O4所制备的λ-MnO2作为正极,在13.6 mA·g−1的电流密度下实现了545.6 mAh·g−1的高放电容量,表明λ-MnO2材料可允许一定量的Zn2+嵌入-脱嵌。
2.5 δ-MnO2
δ-MnO2具有层间距较大的二维层状结构(~0.7 nm),其由共角的MnO6八面体构成,属于单斜晶系,P2/m空间群21。Alfaruqi等42研究了Zn2+在层状的纳米δ-MnO2中的充放电行为,发现Zn2+嵌入时δ-MnO2会转变成尖晶石型ZnMn2O4和层状δ-ZnxMnO2,证实了δ-MnO2在循环过程中会经历相的转变。之后,Jin等43证明了在Zn(TFSI)2电解液中,首次快速充电时Zn2+会嵌入δ-MnO2中,但并不会引起明显的相变。而后续的反应过程是由有H+参与的转换反应主导,这种机制实现了电池稳定的高容量(4000次循环后仍有93%的容量保持率)。此外,δ-MnO2结构的质子吸附能较低,可允许H+与Zn2+共嵌入电极结构中,同时其较大的层间距可满足Zn2+更快的扩散和储存,被作为电极材料广泛应用44。其他隧道结构的MnO2(如α-,β-,γ-和λ-MnO2)在循环时会发生由隧道结构向层状结构的转变,影响电池的稳定性29,37,41,45,而δ-MnO2这种从“层状到层状”的转变在动力学上更有利于Zn2+的存储46。
2.6 R-MnO2
R-MnO2的[1 × 2]隧道框架由共享角的双链结构构成,而双链结构由共享边缘的MnO6八面体组成,属于一种蓝宝石结构47。R-MnO2具有较宽的隧道结构,可满足载流子快速嵌入-脱嵌,并实现较高的电池容量48。
3 二氧化锰的反应机制
MnO2的储能机理非常复杂,目前对于其电化学反应机制仍然存在争议49。根据目前的研究进展,本文总结了以下四种不同的MnO2储能机制:(1) Zn2+的嵌入机制;(2)共嵌入机制;(3) H+嵌入引发的转化反应机制;(4)溶解-沉积机制。对于前两种机制,不同晶型的MnO2在经历Zn2+嵌入时会发生不同的电化学反应和结构转变,且H+作为微酸水系电解液中的组分,理论上可与Zn2+共同参与离子反应。这两种机制均表现出相似的充放电曲线和电压平台。而转换反应机制是一种H+参与的可逆电化学反应实现充放电的储能机制,Zn2+无需嵌入到MnO2隧道中,而是以化合物的形式在MnO2表面生成。至于溶解-沉积机制,其主要是基于在相对高电压区发生的氧化还原反应,利用MnO2可逆的溶解/沉积进行储能,与电解液的pH值有关50。对于嵌入-脱嵌储能机理,电池容量衰减的主要原因是循环过程中发生活性材料的溶解、严重的相变和结构的崩塌,而溶解-沉积机制所使用的酸性电解质会加速锌负极的腐蚀和析氢反应,且发生H+或Zn2+的嵌入会降低电池能量效率。不同晶体结构的MnO2所发生的基本反应不同,其内在复杂机理仍需要继续探索。
3.1 Zn2+的嵌入机制
Zn2+的嵌入机制是Zn2+在正极材料中发生可逆的嵌入-脱嵌进行储能。在弱酸性的Zn(NO3)2水系电解液中,放电时,Zn会快速溶解到电解液中形成Zn2+,此时溶液中的Zn2+会嵌入α-MnO2的隧道中形成ZnMn2O4(Zn2++ 2e−+ 2MnO2→ Zn Mn2O4),同时Mn会经历由+4价到+3价氧化还原态的转变;充电时为放电反应的逆过程,即Zn2+从正极中脱嵌,并重新沉积到负极上29。但不同晶体结构的MnO2在Zn2+嵌入-脱嵌时发生的结构转变类型不同,其反应过程有很大的差异。如,Zhang等45所报道的以Zn(CF3SO3)2和Mn(CF3SO3)2作为电解液的水系锌锰电池(图2a),在第一次放电过程中,Zn2+会嵌入β-MnO2隧道中,经历不可逆相变形成层状Zn-buserite (B-ZnxMnO2·nH2O),随后Zn2+会在该结构内进行可逆的嵌入-脱嵌。γ-MnO2在Zn2+嵌入过程中也会发生相的转变,依次转变成尖晶石型ZnMn2O4、隧道型γ-ZnxMnO2和层状LZnxMnO2,同时伴随锰还原,分别从Mn(IV)态还原成Mn(III)和Mn(II)态,在Zn2+完全嵌入阶段,ZnMn2O4、γ-ZnxMnO2和L-ZnxMnO2共存(图2b)37。需要注意的是,这些在循环过程中发生的相结构转变可能是导致电极结构崩塌和容量衰减的主要因素51。
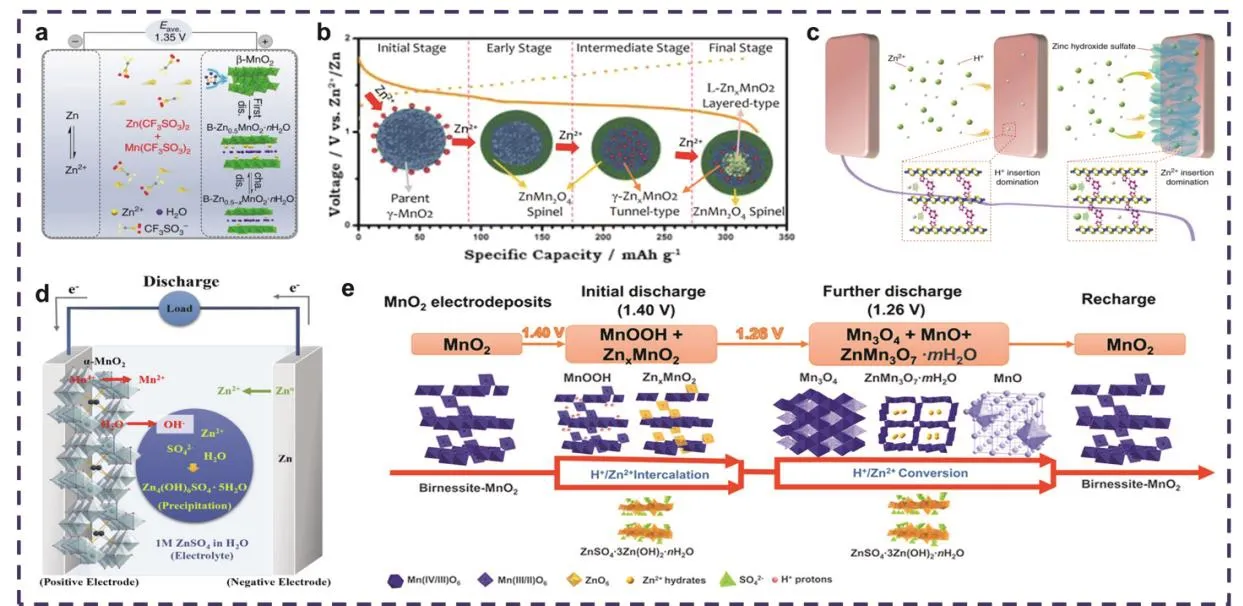
图2 (a)基于CF3电解液的可充电锌锰电池反应机理示意图45;(b) Zn2+嵌入γ-MnO2正极的反应路径示意图37;(c)在层状MnO2中共嵌入H+/Zn2+示意图54;(d)在ZnSO4电解液中Zn//α-MnO2电池放电过程示意图57;(e)在Zn|0.2 mol·L−1 MnSO4(aq),1 mol·L−1 ZnSO4(aq)|Carbon black电池中发生的氧化还原反应及相应化合物结构转变示意图44Fig. 2 (a) Schematic diagram of the rechargeable Zn//MnO2 battery using CF3-based electrolyte 45; (b) Schematic diagram of the reaction pathway of Zn-insertion in γ-MnO2 cathode 37; (c) Schematic diagram of co-insertion H+/Zn2+ in layered MnO2 54; (d) Schematic diagram of showing the reactions during the discharge process for Zn//α-MnO2 battery using aqueous ZnSO4 electrolyte 57. (e) Schematic diagram of the redox reactions and crystal structures of related compounds in the Zn|0.2 mol·L−1 MnSO4 (aq), 1 mol·L−1 ZnSO4 (aq)| carbon black battery 44.
3.2 共嵌入机制
H+的离子半径较小,与Zn2+之间的静电相互作用较弱,且H+作为微酸水系电解液中的关键组分,理论上可参与离子的嵌入-脱嵌反应。Sun等52报道了H+和Zn2+共嵌入储能机制,证实了第一个放电平台(~1.4 V vs. Zn2+/Zn)和第二个放电平台(~1.3 V vs. Zn2+/Zn)分别与H+的嵌入和Zn2+的嵌入有关,同时,放电过程中副产物MnOOH和ZnMn2O4的出现再次印证了H+和Zn2+的共嵌入。值得一提的是,由于H+和Zn2+存在动力学和热力学上的差异,有报道称H+和Zn2+的共嵌入比单一嵌入Zn2+具有更大的离子扩散速度和更低的嵌入电阻,即水系锌锰二次电池更倾向于有H+参与的共嵌入53。同时,Huang等54报道了一种有“自我调节机制”的共嵌入反应(图2c),在初始放电阶段,主要发生H+的连续嵌入,这会提高溶液中OH−的局部浓度,但尚不足以形成片状Zn4(OH)6SO4·nH2O(ZHS)。随着二氧化锰表面的H+浓度持续降低,电池的放电平台降低,此时主要发生Zn2+的嵌入,溶液中的OH-则会与Zn2−、和水发生反应,诱导ZHS在正极表面形成。Zn2+和H+的同时嵌入会进一步促进片状ZHS的形成。需要注意的是,ZHS的形成会消耗多余的OH−,平衡溶液pH值,这有利于提高电池的循环稳定性。此外,Li等44阐明了在不同电压下Zn//MnO2电池发生的氧化还原反应和化合物的晶体结构转变(图2e),初始放电到1.40 V时,发生H+/Zn2+共嵌入反应,MnO2会转变成MnOOH和Zn0.125MnO2,当放电到1.26 V时,则发生H+/Zn2+转换反应,MnOOH和Zn0.125MnO2会进一步转变成Mn3O4、MnO和ZnMn3O7·2H2O,充电时又重新恢复成MnO2,需要注意的是,由于反应体系的复杂性,在反应过程发生一些难以避免的副反应,导致ZnSO4·3Zn(OH)2·nH2O副产物的生成。
近期Wang等55提出了一种H+/NH4+共嵌入机制,即H+和NH4+可以同时嵌入到MnO2隧道中。嵌入的H+会与MnO2中的O2−结合形成O-H键,O-H键与Mn-O键的竞争效应会增大后者氧原子的电子密度,而NH4+的嵌入会增强NH4+与MnO2之间的氢键,从而增大Mn3+O6八面体结构的稳定性。同时,H+和NH4+之间的协同作用可有效地提高载流子的扩散动力学。根据Bader电荷分析,H+/NH4+共嵌入状态下的电荷转移数小于仅嵌入H+或NH4+,并经计算发现,共嵌入的扩散能垒约为0.37 eV,低于仅嵌入H+或NH4+(0.51或0.43 eV)。这种共嵌入机制有效提高了锌锰电池的电化学性能,实现了在4 A·g−1的电流密度下经过4000次循环后仍能保持93.3%的容量。然而,目前MnO2中载流子的反应路径仍存在争议,研究策略不够全面,需要对不同晶型的MnO2的质子反应机制做出更加全面系统的研究。
3.3 转化反应机制
与传统的Zn2+嵌入/脱出机制不同,Pan等56报道了一种电解液中的H+参与α-MnO2电化学反应的机制。在弱酸性的ZnSO4+MnSO4电解液中,α-MnO2会与水中的H+形成MnOOH,而为了平衡溶液电荷,在放电时多余的OH-会与ZnSO4和H2O反应,在正极表面生成ZnSO4[Zn(OH)2]3·xH2O。充电时,ZnSO4[Zn(OH)2]3·xH2O会逐渐溶解,同时MnOOH也会恢复为原始的MnO2。其正负极的电极反应情况可由下式表示:
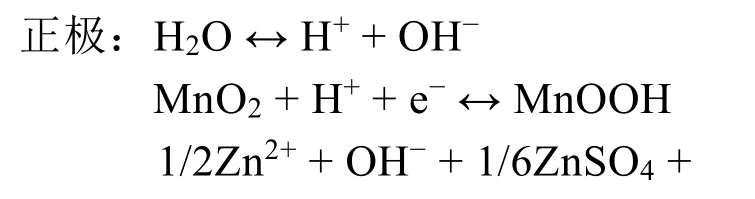
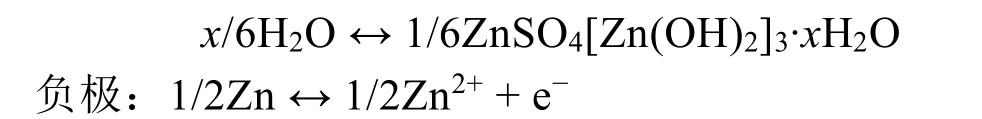
基于上述讨论,转化反应理论上与MnO2的晶体结构和电解液的条件(如离子浓度)有关。随后,Lee等57通过研究循环过程中离子的传输与反应行为,发现在放电过程中锰会发生歧化溶解到电解液中,提高了电解液的pH值,导致Zn2+会以Zn4(OH)6(SO4)·5H2O的形式在α-MnO2表面沉积(图2d)。但该化合物可以通过酸洗去除,说明Zn2+并未嵌入到α-MnO2隧道中,这与传统观念中的Zn2+嵌入机制不同。之后,Yang等58通过对放电后的正极形态和结构进行研究,证明了电池的放电过程为α-MnO2和MnOOH之间的转化反应,无需Zn2+嵌入隧道中形成ZnMn2O4或ZnxMnO2。值得一提的是,目前对转换反应机理的研究不多,相关报道较少,仍以传统的嵌入-脱出为主。
3.4 溶解-沉积机制
相比于传统的嵌入-脱嵌机制在Mn4+/Mn3+氧化还原过程中只利用了1个有效电子转移,基于2个有效电子转移数的溶解-沉积机制有效提高了电池的比容量和输出电压,其理论容量和理论电压分别可达616 mAh·g−1和1.991 V vs. Zn2+/Zn59,60。根据方程2可知,溶解-沉积机制与溶液的pH值密切相关,MnO2/Mn2+之间的转化会随着pH值的降低而逐渐加强61。Chao等62通过添加H2SO4将电解液pH值降低至1左右,实现了MnO2/Mn2+较为充分的可逆转化(图3a)。在充电过程中,游离的Mn2+在正极集流体表面失去两个电子转变成了二氧化锰,在这个过程中会不断地生成H+。而在放电时,正极表面的二氧化锰获得两个电子重新转变为Mn2+。整个反应过程如下述方程所示:
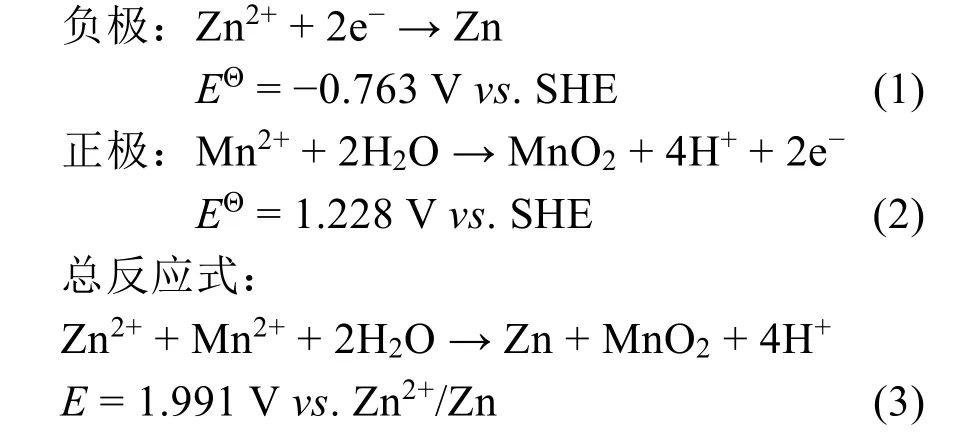
得益于这种独特的机理,所制备的Zn//MnO2电池在高酸度环境下实现了很高的比容量(~570 mAh·g−1)、优异的输出电压(~1.95 V vs. Zn2+/Zn)和卓越的能量密度(1100 Wh·kg−1) (图3b)。
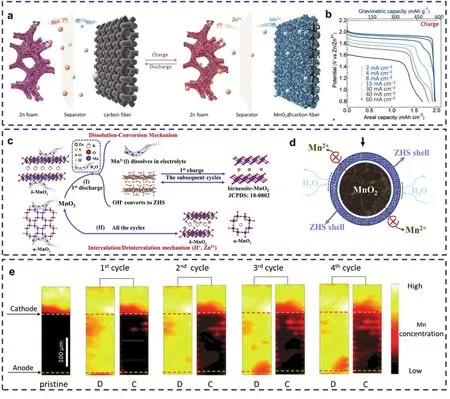
图3 (a)基于溶解-沉积机制的锌电池示意图以及(b)其恒流放电曲线62;(c)基于溶解-沉积机制和嵌入-脱嵌机制的锌电池示意图以及(d)ZHS在放电过程中的作用机制50;(e)完全放电(D)和充电(C)状态下的Mn荧光图63Fig. 3 (a) Schematic diagram of zinc battery based on dissolution-deposition mechanism and (b) its Galvanostatic discharge curves 62. (c) Schematic diagram of zinc battery based on dissolution-deposition mechanism and intercalation mechanism and (d) the role of ZHS in the discharge process 50. (e) Mn fluorescence maps at fully discharged (D) and charged (C) states 63.
这种溶解-沉积储能机制除了在酸性电解液中有过报道外,近期有研究人员也在弱酸性电解液中观察到了二氧化锰的溶解-沉积过程。如Guo等50认为,Zn//MnO2电池在首次放电过程中,MnO2与H2O反应生成Mn2+和OH−而提高电解液的pH值,此时OH−会立即与周围的ZnSO4发生反应生成Zn4(OH)6SO4·4H2O;充电时Zn4(OH)6SO4·4H2O则会与Mn2+反应生成bimeessite-MnO2,后续的充放电过程为bimeessite-MnO2的溶解-沉积过程(图3c),此过程即为Zn//MnO2电池的放/充电过程。有趣的是,Zn4(OH)6SO4·4H2O会通过控制活性水的方式抑制Mn的溶解沉积行为(图3d),但同时又会消耗掉电解液中多余的OH−,促进循环反应的持续进行,其复杂的作用机制仍待探索。值得一提的是,H+/Zn2+的嵌入-脱出仍会发生在未充分溶解的MnO2中,只是对容量的贡献值较小。同时,Wu等63利用X射线荧光映射,分析了在弱酸性电解液中锰基的具体溶解-沉积行为(图3e)。放电时,MnO2正极会溶解成Mn2+,而充电时,MnO2则会重新沉积在正极上,但同时可观察到一种嵌锌的层状锰化物(ZnMn3O7·3H2O)的存在。这种MnO2的溶解-沉积和H+/Zn2+的嵌入-脱嵌出共存的现象说明了这两种机制可能普遍共同存在于水系锌二次电池体系,只是对于容量的贡献值不同。
目前MnO2的储能机制主要以离子的嵌入-脱嵌和锰基的溶解-沉积机制为主。以电荷载流子在正极材料中嵌入-脱嵌进行充放电循环是锌锰二次电池典型的储能机制,其有效转移电子数为1,电解液通常为弱酸性。在充放电过程中发生的活性材料溶解、不可逆相变以及相变过程中造成的结构崩塌是导致这类机制容量快速衰减的主要原因64。尽管对于二氧化锰的储能机理已经研究多年,但到目前为止,由于水系电池体系中复杂的pH值变化和存在各种副反应等问题,隧道型、层状和尖晶石状等不同晶体相的MnO2的基本反应机制仍不明晰,对其充放电行为的理解仍存在争议,需要进行进一步全面且统一的研究。此外,一种MnO2与电解液中的H+之间发生转化反应的储能机制被提出,化合物的生成-溶解即为电池的充放电过程,其与传统的Zn2+嵌入-脱嵌MnO2隧道实现电池循环的观念不同56。但目前对于这种机制的报道较少,有待更加深入的研究。近期,不断有学者提出了具有2个有效转移电子数的MnO2溶解-沉积储能机制,并成功应用于水系锌锰二次电池中,该机制深受电解液中pH值的影响61,通常需要酸性电解液来实现充分的溶解/沉积反应。H+的浓度在电化学反应中起到至关重要的作用。相比于传统的嵌入-脱嵌机制,溶解-沉积储能机制合理地利用了难以避免的锰溶解的问题。值得一提的是,Xue等49指出电解液添加剂MnSO4的作用可能并不是抑制MnO2的溶解,而是促进MnO2/Mn2+更充分可逆的沉积-溶解,从而提高电池的循环稳定性和比容量。实际上,嵌入-脱出机制与溶解-沉积机制在水系锌锰二次电池中应该都是普遍存在的,但由于两种机制的转移电子数不同,在基于溶解-沉积机制的电池体系中发生H+/Zn2+的嵌入可能会降低电池的比容量。因此,如何明晰这两种机制的反应路径,并有效地结合两者的优势,突破现有容量瓶颈,开发出高性能水系锌锰二次电池,是目前所需要深入研究的问题。
4 二氧化锰的优化措施
目前,对于嵌入-脱嵌机制,需要解决循环过程中发生的活性材料溶解、本征电导率低、不可逆相变和结构崩塌,以及由于Zn2+与宿主材料之间的强静电相互作用而导致的离子扩散动力学行为变差,和电极添加剂(如PVDF)的使用会阻碍载流子的传输等问题。而溶解-沉积机制当前仍处于探索阶段,其内在机理并未明晰。在该体系下电池的高容量和高电压平台依赖于充分可逆的锰溶解。此外,反应过程中发生的不可避免的H+/Zn2+嵌入以及强酸性的电解液会加速金属锌负极的腐蚀和析氢等问题,都会造成Zn//MnO2电池较差的循环稳定性和库伦效率。为了有效提高Zn//MnO2电池的各项电化学性能,下文将针对MnO2主要的储能机制(嵌入-脱嵌和溶解-沉积机制),总结MnO2正极材料现有的优化策略。
4.1 针对嵌入-脱嵌机制的优化策略
4.1.1 复合材料改性
复合材料改性策略可通过结合碳基材料、有机物等来提高电极材料导电性,同时提供更大的比表面积和更多的活性位点,并有效防止相变造成的结构崩塌,进而提升水系锌锰二次电池电化学性能64。
常用于复合的碳材料有碳纤维、石墨烯、碳纳米管等。其中碳纤维具有特殊的离子/电子传输通道和收集定向电子的功能,且对于水介质具有较低的电化学活性和较宽的电化学窗口。同时碳纤维还可起到支撑作用,防止发生结构崩塌,提高电池的循环稳定性。Chen等65合成了一种在碳纳米纤维(CNFs)表面原位生长出的垂直排列的MnO2纳米片结构(MOC,图4a),导电的CNFs与MnO2的紧密联系形成的互连网络显著提高了正极材料的离子/电子转移动力学,且层间的结晶水起到了结构支架的作用,维持了活性材料内部结构的稳定。同时,超薄的MnO2纳米片增大了电极与电解液界面的接触面积,有效增加了反应活性位点。此外,通过测定由不同KMnO4/CN质量比F (0,5 : 1,15 : 1)制备的MOC在不同电流密度下的比容量(图4b),发现其中MOC-5在200 mA·g−1电流密度下实现了297 mAh·g−1的高放电容量,且当电流密度从3000 mA·g−1回到200 mA·g−1后,容量仍能保持在283 mAh·g−1,表现出了高达95%的容量保持率,同时其在200 mA·g−1的电流密度下经过700次循环后仍保留有221 mAh·g−1的高容量(图4c)。碳布是一种重量轻、抗弯折能力强、电导率高和化学稳定性强的材料,在其表面电沉积MnO2可以制备出高性能电极材料,有效提高离子/电子的传输速率,表现出良好的导电性和电化学稳定性66。除了碳布外,经过优化改性制备的碳材料能够显著提高自身的物理化学性能,可应用于改善电池性能。如氮掺杂碳纳米片(N-CNSs)网络由具有丰富的纳米孔结构的N-CNS相互连接而成,该多孔骨架框架能增大材料比表面积,并提供更多的离子/电子传输通道,同时掺杂N时所形成的缺陷能够有效提高反应活性,是良好的导电材料67。Fu等68通过金属-有机框架模板策略制备出了具有多孔骨架的氮掺杂锰基正极材料(MnOx@N-C),得益于材料的多孔性、独特渗氮导电碳网络结构和电解质中Zn2+和Mn2+的协同作用,其表现出了优良的电化学性能。同时,还可利用三维多孔泡沫骨架具有高电导率的优势以提高电子转移和离子扩散速率,如一种由α-MnO2和具有三维多孔泡沫结构的碳纳米管(CNTs)复合而成的薄膜材料α-MnO2@CNT,其能有效改善离子/电子扩散动力学,赋予了电极材料更快的离子扩散速率和更优异的导电性,在0.97C(1C = 308 mA·g−1)的 电 流密度 下 实现了 308.5 mAh·g−1的放电比容量69。
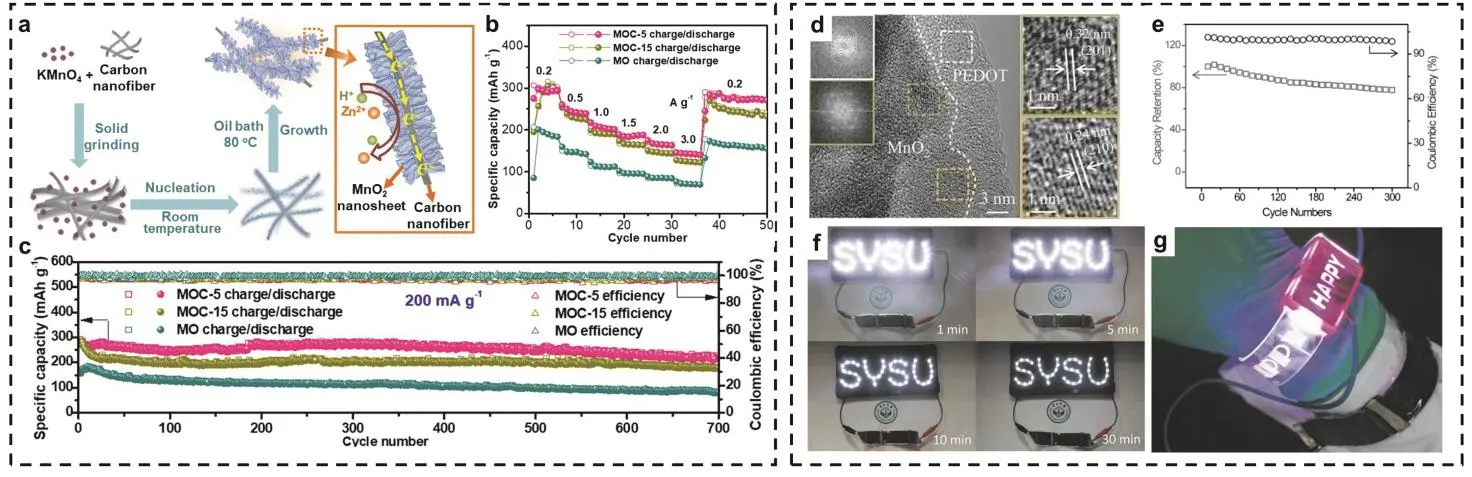
图4 (a) MOC的合成示意图;(b)以MOC-5,MOC-15和MO分别作为正极,2 mol·L−1 ZnSO4和0.1 mol·L−1 MnSO4作为电解液的的锌离子电池在不同电流密度下的倍率性能以及(c)在0.2 A·g−1电流密度下的循环性能65;(d) MnO2@PEDOT样品的高分辨透射电镜(HRTEM)图像以及(e)柔性准固态Zn//MnO2@PEDOT电池在1.86 A·g−1电流密度下经过300次循环后的循环性能和库伦效率;(f)三个柔性准固态Zn//MnO2@PEDOT电池供电的由45个发光二极管组成的霓虹灯以及(g)电池为手表上的LED灯供电的图片72Fig. 4 (a) Schematic diagram of the synthetic procedure of MOC. (b) The rate performance of zinc ion battery with MOC-5, MOC-15 and MO as positive electrodes and 2 mol·L−1 ZnSO4 and 0.1 mol·L−1 MnSO4 as electrolyte at different current densities and (c) the cycle performance at current density of 0.2 A·g−1 65. (d) HRTEM images of MnO2@PEDOT sample and (e) Cycling performance and Coulombic efficiency collected at 1.86 A·g−1 for 300 cycles of flexible quasi-solidstate Zn//MnO2@PEDOT battery. (f) Photographs of a neon sign composed of 45 light-emitting diodes powered by three flexible quasi solid Zn// MnO2@PEDOT battery devices and (g) a watch with LED lights powered by three devices 72.
正极表面的涂层可形成稳定的固体电解质界面(简称SEI膜),能有效抑制Mn2+的溶解,增强电极表面稳定性,从而提高电池循环性能。已报道的涂层材料可分为有机物和无机物两大类。作为最典型的例子,Wu等70提出了一种石墨烯(rGO)均匀包覆的α-MnO2(MGS)正极材料。利用电感耦合等离子体(ICP)分析在2 mol·L−1ZnSO4电解液中锰的溶解行为,发现在放电过程中,Zn//MGS电池电解液中Mn的浓度比在原始Zn//MnO2电池中要低,说明rGO涂层的存在有效抑制了电池循环过程中MnO2的溶解。因此,Zn//MGS电池能在0.3 A·g−1条件下经过100次循环后仍具有406.6 Wh·kg−1(382.2 mAh·g−1)的高能量密度。但石墨烯的高昂成本也大大限制了其大规模商业化的发展。除了石墨烯,其他碳材料如无定形碳等,其独有的多孔性不仅能有效提高离子/电子的传输速率,还能建立稳定的电极结构,实现优异的电化学性能68。此外,研究人员还报道了利用原位电化学法在二氧化锰表面形成CaSO4·2H2O的SEI薄膜保护层的策略,有效提高电池电化学稳定性,延长使用寿命71。需要注意的是,由于非导电涂层会降低载流子的扩散速率,在设计时应考虑正极界面处离子传输和电解液可及性的问题。
导电聚合物不仅具有较高的导电性、优异的化学稳定性和较强的电荷储存能力的优势,还具有环境友好、成本低廉等特性,可作为活性材料的理想保护涂层。Zeng等72报道了一种用于修饰MnO2界面的聚3,4-乙基二氧硫吩(PEDOT)涂层(图4d),PEDOT缓冲涂层在抑制锰溶解和结构崩塌的同时保证了较高的载流子传输速率。同时,MnO2@PEDOT可直接在柔性碳布上制备,避免使用粘结剂。以PVA/ZnCl2/MnSO4凝胶作为电解液,所制备出的柔性准固态电池Zn//MnO2@PEDOT具有良好的循环性能和库伦效率(图4e),可为发光二极管供电(图4f),并作为柔性电源为手表供电(图4g),表明其在可穿戴电子产品中具有广阔的应用前景。此外,有研究人员提出聚吡咯(PPy)和聚苯胺(PANI)等导电聚合物也可以充当二氧化锰的涂层以缓解在充放电过程中活性材料的溶解73,74。
4.1.2 纳米结构优化
苏:豪迈!羌族沙朗舞的这种感染力让人心里很舒服,就会使我情不自禁地加入当中去。在各种丰收、节庆的日子里,不论男女老少都要跳。沙朗舞的风格比较淳朴,节奏比较轻快,动作比较粗犷,具有强烈的民族艺术感染力,容易吸引大家的注意力和兴趣,充分显示出我们的羌族文化气息及独特的艺术风格。
纳米尺寸的电极材料具有较大的表面积和优异的尺寸效应(包括微小尺寸效应与真实尺寸效应)。纳米材料有更大的电解液接触面积,载流子容易在纳米结构表面扩散,这显著提高了电池的倍率性能。此外,纳米材料还可缓解体积膨胀问题,维持结构稳定,从而实现长循环寿命75。根据公式τeq= L2/2D (τeq为扩散时间,L为材料尺寸,D为扩散系数)可知,合成纳米尺寸的电极材料(即降低L)可有效降低离子的τeq,从而提高电池充放电速率。
MnO2的晶体形态很大程度上决定Zn2+的存储性能22,目前,不同晶体结构和形貌的纳米MnO2在水系锌锰二次电池中得到广泛的研究。Kim等76报道的3D分层介孔γ-MnO2纳米球(UW-MO)结构具有比本体γ-MnO2更优异的循环稳定性和电荷容量,在50个循环后UW-MO仍有400 mAh·g−1的比容量,而MO逐渐下降到180 mAh·g−1。此外,与其他隧道型的MnO2晶型相比,β-MnO2具有较窄的隧道结构,不利于Zn2+的存储。而Lslam等30采用微波辅助水热合成法制备了暴露(101)平面的β-MnO2纳米棒,并将其作为水系锌锰二次电池的正极材料(图5a),其中,(101)峰在Zn2+嵌入β-MnO2后会向更低的角度转变,这归因于锰的还原导致Mn―O键的延伸(图5b)。Zn2+嵌入-脱嵌过程中,β-MnO2纳米棒电极分别在0.15、0.25和0.3 nm处表现出三个不同的傅里叶变化(FT)峰,分别对应Mn―O,共享边缘的Mn―Mn和共享角的Mn―Mn。将β-MnO2纳米棒沿a轴旋转约90°后,可以看到纳米棒中的[1 ×1]开放隧道,这种独特的一维棒状形态增大了电极与电解液的接触面积,有利于Zn2+在β-MnO2结构内的嵌入-脱嵌。所制备的电池在100 mA·g−1电流密度下实现了高达270 mAh·g−1的放电比容量,以及经过200次循环后仍保持75%的容量保持率和100%的库伦效率(200 mA·g−1电流密度)。纳米结构的优异性还体现在其他晶型当中,如Liu等77提出的具有(1D-3D)混合网络结构的δ-MnO2纳米线正极,这种独特的结构增大了电极与电解质的接触面积,提供了更多活性位点和快速的离子传输通道。以及Alfaruqi等78报道的一种纳米α-MnO2正极,其具有优异的结构稳定性,显著提高了Zn2+嵌入正极的可逆性。
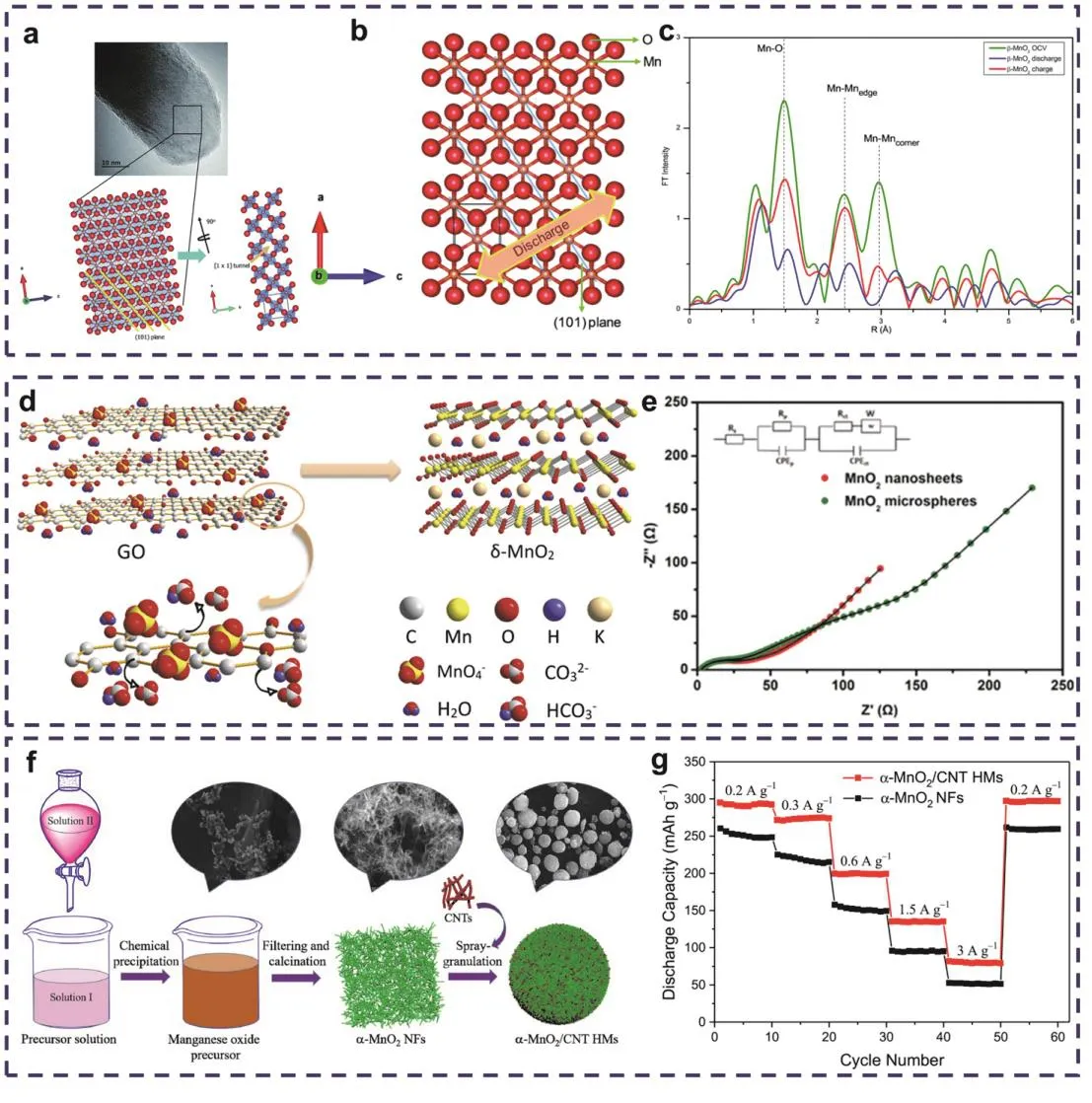
图5 (a) β-MnO2纳米棒样品的晶体结构以及(b) β-MnO2结构内(101)晶格平面示意图;(c) Zn//MnO2经历充放电后β-MnO2纳米棒的EXAFS光谱30;(d) δ-MnO2纳米片的形成示意图;(e) δ-MnO2纳米片与微球电极的Nyquist示意图(黑线为拟合线) 79;(f)不同合成阶段的α-MnO2 NFs和α-MnO2/CNT HMs的制备示意图和相应产物的SEM图像以及(g)将α-MnO2 NFs和α-MnO2/CNT HMs作为电极制备的锌离子电池(ZIBs)在电压范围为1.2–1.85 V时的速率能力比较81Fig. 5 (a) Schematic diagram of crystal structure of β-MnO2 nanorod sample and (b) (101) lattice planes in the β-MnO2 structure. (c) EXAFS spectra of the β-MnO2 nanorod cathode collected after discharging/charging 30.(d) Schematic diagram of the formation of δ-MnO2 nanosheets. (e) Nyquist diagram of δ-MnO2 nano sheet and microsphere electrode (black line is fitting line) 79. (f) Preparation diagrams of α-MnO2 NFs and α-MnO2/CNT HMs at different synthesis stages and SEM images of corresponding products and (g) rate capability comparison at voltage cutoffs of 1.2–1.85 V of ZIBs with α-MnO2 NFs and α-MnO2/CNT HMs 81.
纳米尺寸的电极材料会缩短离子扩散的距离,缩小扩散时间,改善电化学反应动力学75。Guo等79制备了一种超薄δ-MnO2纳米片结构(图5d),通过对超薄δ-MnO2纳米片和纳米球结构进行电化学阻抗谱测试(EIS) (图5e),可发现具有纳米片结构的δ-MnO2的电荷转移电阻为46 Ω,远小于纳米球δ-MnO2的223 Ω,这可能是因为离子在厚度为2–4 nm的薄纳米片中的扩散路径小于在50 nm厚的纳米球中,因此所需的扩散时间更短。此外,Wang等80所报道的2D单原子层纳米MnO2作为正极会在水平和垂直方向上提供相互交联的离子传输通道,相较于多层纳米MnO2,单层纳米MnO2具有更短的离子扩散路径,可实现更快的电子/离子传输速率。这基本上可以说明更小尺寸的纳米结构具有更快的电化学反应速率。此外,具有纳米尺寸的活性材料更容易适应离子嵌入-脱嵌时所发生的体积变化,有效防止相变造成的结构坍塌,表现出优异的循环稳定性。如Liu等81利用电化学沉积法制备了α-MnO2与纳米纤维/碳纳米管的复合材料(α-MnO2/CNT HMs) (图5f),其独特的复合结构有助于提高电极材料的电化学稳定性。再加上纳米纤维α-MnO2(α-MnO2NFs)与CNTs之间形成了一种紧密排列的网络结构,两者之间的协同作用提供了更多可快速转移电荷的通道和丰富的活性反应位点,有效改善了活性材料的电荷转移动力学,从而提高了电池的倍率性能(图5g)。
需要注意的是,尽管纳米材料具有诸多优势,但仍存在诸如热稳定性下降、表面副反应多等问题75。如纳米颗粒由于具有非常高的比表面积和表面能,会倾向于形成团聚体,导致接触电阻升高,或在循环过程中发生电化学团聚现象,降低电池容量,同时还会增加电极与电解液之间发生副反应的概率。此外,纳米结构的电极材料相比于普通电极具有更窄的电化学稳定窗口。因此,在设计纳米结构改性电极材料时,需综合考虑纳米材料的优劣势来设计高性能锌锰二次电池正极材料。
4.1.3 客体预嵌策略
不可逆的非平衡相变以及嵌入层结构的不稳定性会导致电池容量衰减,影响电池性能82。通过在具有层状结构的MnO2中预嵌入客体,如水分子83、有机分子84和无机金属离子85等,能够为Zn2+/H+的嵌入-脱嵌提供稳定的内部结构,缓解其在嵌入-脱出过程中引起的结构崩塌,优化晶体结构,同时减弱Zn2+离子与MnO2之间的静电相互作用,提高载流子的扩散速率。相比于其他具有隧道结构的MnO2晶型,δ-MnO2具有较宽的层间距,更适合预嵌入客体,是大多数预嵌入策略的选择。

图6 (a) PANI-MnO2纳米层结构示意图;(b)经过400 °C热处理去除PANI后的PANI-MnO2纳米层结构的HR-TEM图像54;(c) cw-MnO2的XRD图案和晶体结构;(d)左图:Zn插层结构和所需的相应能量;右图:锌的球内络合效应导致面向锰的突出,形成放大的Zn-Mn哑铃结构(数字代表原子间的距离) 83;(e)预嵌Na+和水分子的δ-MnO2充放电工作示意图;(f) δ-NMOH电极的非原位XRD图87Fig. 6 (a) Schematic diagram of PANI-MnO2 nanolayers structure. (b) HR-TEM image of PANI-MnO2 nanolayers structure after removing PANI by heat treatment at 400 °C 54. (c) XRD pattern and crystal structure of cw-MnO2.(d) Left: Zn-intercalation structure and corresponding energy; Right: the inner-sphere complexation of zinc leads to the protrusion of the facing Mn, constituting a Zn-Mn dumbbell structure as zoomed (numbers represents the distance between atoms) 83. (e) Schematic diagram of pre-intercalated Na+ and water molecules δ-MnO2 during charging and discharging.(f) ex situ XRD pattern of δ-NMOH electrode 87.
将金属阳离子K+、Ba2+、Na+、Co2+/3+作为客体嵌入MnO2会占据特定的晶格位点,或增大电极的晶格间距,并与Zn2+发生相互作用,从而改变正极的导电性。如Zhang等88提出了一种预嵌La3+的纳米δ-MnO2电极材料(LMO),预嵌的La3+可有效降低Zn2+与MnO2间的相互作用力,改善Zn2+的嵌入-脱嵌动力学行为,使得LMO具有更大的储锌容量、更快的反应速率和可逆的氧化还原反应,同时La3+还能对晶体结构起到支撑作用,防止在循环过程中发生结构的崩塌,最终Zn//MnO2全电池展现了超高的容量密度375.9 Wh·kg−1(100 mA·g−1电流密度)。
虽然客体嵌入MnO2被普遍认为可以扩大层间距、促进载流子的嵌入/脱出,并提高结构稳定性82。然而,有报道指出发现引入的金属阳离子会通过物理阻塞和静电排斥作用阻碍载流子在隧道中的扩散89,说明当隧道被预先填充了不轻易去除的金属阳离子时,可能会大大降低载流子嵌入/脱出速率。因此,在设计客体预嵌改性MnO2正极时,应综合考虑外来离子对于载流子运输的阻碍作用和其对于稳定材料结构的贡献等方面,从而开发出更优异的电极材料。
4.1.4 缺陷工程
电化学反应与电极材料的电子结构有密切的关系,其可决定载流子的扩散速率,而晶体结构中的缺陷会影响电子结构的分布。晶体中缺陷的类型主要有点缺陷(如空位)、线缺陷(如位错)、面缺陷(如晶界)和体缺陷(如杂质引起的晶格无序状态)。缺陷的存在可提供更多载流子的储存/吸附/活性位点,有效减小离子间的静电斥力,克服迁移和扩散障碍,并降低反应势垒,从而提高离子的嵌入/脱出速率90。
在MnO2中掺杂异质元素会导致在原带隙上产生新的缺陷态(如空位),处于缺陷态上的电子结构会发生改变,降低锌嵌入的能垒,从而实现更优异的电化学性能。通过掺杂异质元素所形成的氧空位能有效调控其表面化学和几何构型,如Lian等91报道了一种Ti掺杂α-MnO2,虽然Ti的掺杂会导致层间收缩,但由于锰价态的降低,会产生氧空位进行电子补偿。而氧空位会打开[MnO6]八面体壁,导致晶体结构中电荷分布不均和局部电场不平衡,从而加速离子/电子的扩散速率,实现高倍率性能和长循环稳定性。同时,Fang等92报道了一种K+掺杂与氧缺陷复合材料(K0.8Mn8O16,KMO)。与原始的α-MnO2相比,KMO具有更快的H+扩散速度,有效提高了电化学反应动力学(图7a)。这是因为氧缺陷能有效降低氧化还原反应过程中电子运输和电荷转移的能量势垒,并打开了α-MnO2中[MnO6]多面体壁,促进H+的扩散(图7b)。所制备的Zn//K0.8Mn8O16电池实现了398 Wh·kg−1的高能量密度,在经过1000次充放电循环后仍无明显的容量衰减(图7c),显示出了缺陷工程在发展高性能正极材料方向上的潜力。此外,Kim等31报道了一种掺杂氟(F)的β-MnO2交错球状纳米片正极材料,这种F掺杂策略有效扩大了β-MnO2的晶格尺寸,提供了更多的电化学反应活性位点。同时因掺杂而引入的氧缺陷不仅会通过改变局部电子结构来提高电导率,还能降低锌嵌入的能量势垒,进而促进锌离子的嵌入/脱嵌行为。结合交错稳定的内部网络结构,实现了优异的循环稳定性和倍率性能,在1800 W·kg−1功率密度下实现158 Wh·kg−1的高能量密度,且在经过150次循环后容量保持率仍达85%。
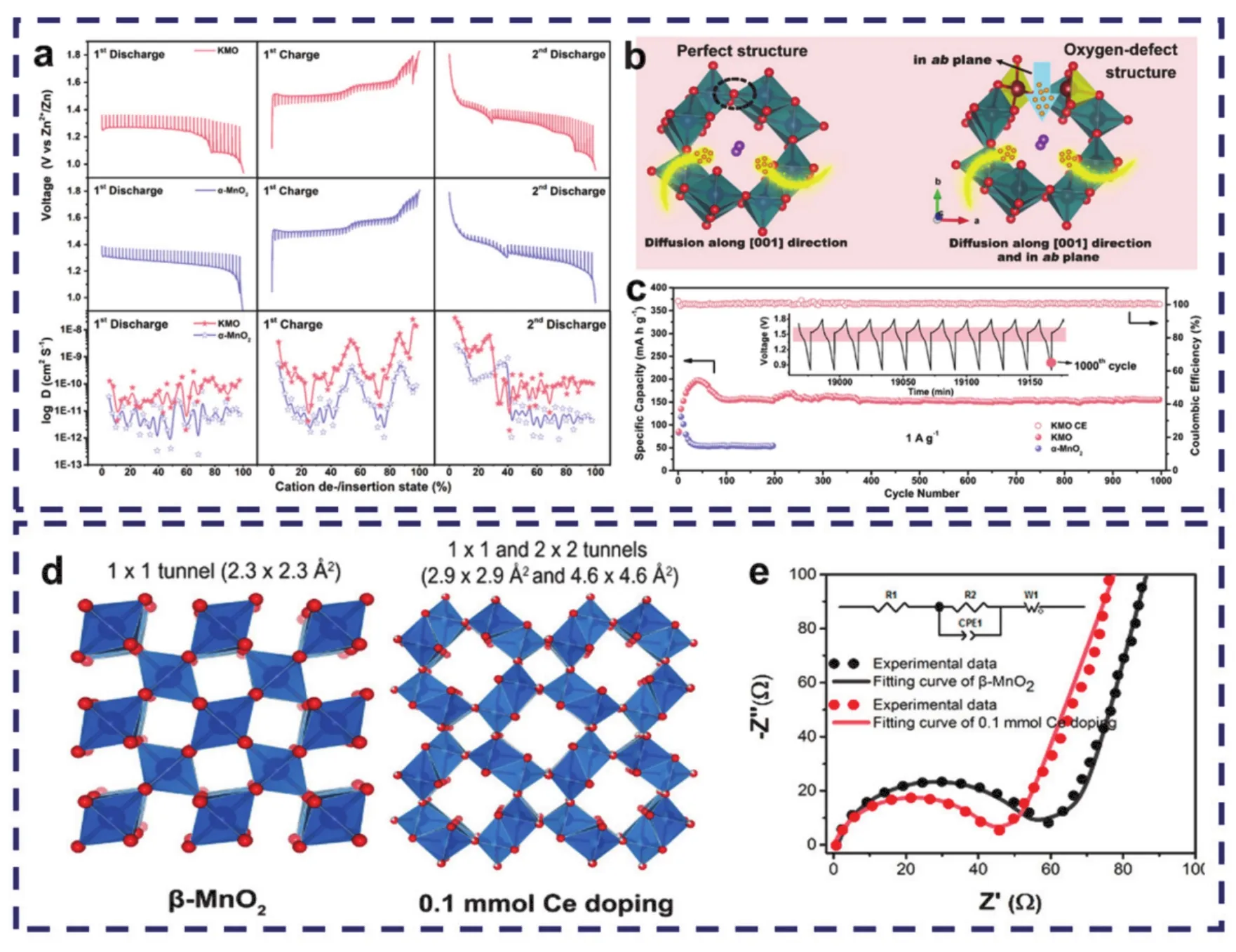
图7 (a)KMO和α-MnO2电极在不同放电/充电状态下的GITT曲线和相应的H+扩散系数;(b) H+在KMO的完整结构及含氧缺陷结构中扩散示意图;(c) KMO和α-MnO2电极在1000 mA·g−1下的长寿命循环性能和KMO电极的最后十条充放电曲线92;(d) β-MnO2和Ce掺杂β-MnO2的晶体结构;(e) β-MnO2和0.1 mmol Ce掺杂β-MnO2的EIS奈奎斯特图和等效电路85Fig. 7 (a) GITT curves and corresponding H+ diffusion coefficients of KMO and α-MnO2 electrodes under different discharge/charge states. (b) Schematic diagram of H+ diffusion in the complete structure and oxygen defect structure of KMO. (c) Long cycle performance of KMO and α-MnO2 electrode and the last ten charge/discharge curves of KMO electrode at 1000 mA·g−1 92. (d) Crystal structure of β-MnO2 and Ce doping β-MnO2;(e) EIS Nyquist diagram and equivalent circuit of the β-MnO2 and 0.1 mmol Ce doping cathode 85.
4.1.5 元素掺杂
异质元素与MnO2具有不同的电子构型,可以调节材料的表面形态,诱导结构发生转变,极大地影响电极材料的结构和电化学性能。此外,利用Mn与异质元素的键合作用,可增强晶体结构稳定性,防止其在相变过程中发生坍塌,提高MnO2正极的电荷存储性能93。
稀土元素具有特殊的4f电子构型,可与MnO2相互作用,调节其表面形态。结合纳米结构与金属元素掺杂的优异性,Wang等85提出了一种通过水热法合成的Ce掺杂MnO2纳米棒正极材料,Ce的掺杂诱导MnO2从原来的β相转变成新的α相(图7d),出现了[2 × 2]隧道结构。其循环伏安(CV)曲线表明,Ce掺杂β-MnO2的正极峰和负极峰中心之间的电压差明显小于未掺杂的β-MnO2。且Ce掺杂β-MnO2的电荷转移电阻远低于本体β-MnO2,表明前者具有更优异的导电性(图7e)。基于改性β-MnO2制备的Zn//MnO2电池能在5C (1C = 308 mA·g−1)的电流密度下实现134 mAh·g−1的高容量。除了稀土元素外,Alfaruqi等94报道了一种利用氧化还原反应制备的V掺杂MnO2纳米正极,这种V掺杂MnO2的平均晶粒尺寸小于原始的MnO2,增大了活性材料的比表面积,且增加了反应活性位点,有效提高了材料的导电性。相比于原始MnO2,其具有更高的放电容量(266 vs. 213 mAh·g-1)。此外,掺杂微量元素(如N、La、Ca等)也可以降低电荷转移电阻,从而改善MnO2的导电性,进而提升电池的电化学性能95,96。
氟掺杂是一种常见的改善电极电化学储能的方法98。它不仅可以诱导缺陷的生成和提供更多的Zn2+电化学活性位点,其极高的电负性和对离子的吸附能力还能有效调节电极性能,从而促进Zn2+的嵌入/脱嵌行为。同时,Liu等93还提出了一种Mn-F键的引起的“钉扎效应”,这种效应构建了一种活性高且稳定的晶格框架,并提供快速的离子传输通道,在0.6C (1C = 308 mA·g−1)的电流密度下实现了高达311.6 mAh·g−1的比容量,以及在5C下经过1200次循环后仍无明显的容量衰减。值得一提的是,氧具有高电负性,采用低电负性元素如S和C等取代氧从而降低Zn2+在晶体结构中的扩散势垒似乎可以实现。但目前少有对阴离子掺杂的研究报道,有待进一步研究。
4.2 针对溶解-沉积机制的优化措施
近年来,溶解-沉积机制由于其高放电电压、高能量密度等优势受到了研究人员的大量关注。Chao等62在水系锌锰二次电池中提出了这种具有双电子转移的氧化还原反应,基于此机制所制备的Zn//MnO2电池的理论容量可达616 mAh·g−1,这是单电子氧化还原反应容量(308 mAh·g−1)的两倍,同时,其理论电压可达1.991 V vs. Zn2+/Zn,能量密度可超过1100 Wh·kg−1。当前,基于溶解-沉积机制已经能成功制备出接近理论容量的高容量电池。但仍存在反应路径尚未明晰(如无法明确放电平台降低的原因)、酸性的电解液会对负极有腐蚀作用以及MnO2难以充分溶解等问题。目前所报道的优化措施主要有调节电解液成分、活性催化以及制备解耦体系、液流体系电池等。
通过改变电解液成分,可有效调控MnO2的溶解沉积行为,并控制锌负极腐蚀和析氢等问题。Zeng等98报道了一种以接近中性的Zn(CH3COO)2和Mn(CH3COO)2作为电解液的Zn//MnO2电池(图8a)。CH3COO−是一种典型的弱酸离子,具有强极化性和高电负性。电解液中的CH3COO−会吸附在MnO2表面的Mn位点,这显著削弱了其与水的结合作用,改变了MnO2正极的表面性质,进而降低Mn溶解的能量势垒(图8b)。同时,接近中性的电解液可抑制锌负极析氢和腐蚀的问题,并提供了556 mAh·g−1的高比容量和1.5 V的工作电压。此外,Lei等99提出了一种碘化物(I−)介质调节策略,即I−会将固态的MnO2还原成Mn2+,同时自身被氧化成I3−,并在后续的充电过程中重新还原成I−。这种I3−/I−对MnO2//Mn2+的协同作用可提高未充分溶解的MnO2的活性,使其具备循环溶解-沉积的能力。同时,Zheng等100报道了一种溴(Br−)介质调节的Zn-Br2//MnO2电池,这种溴介质可以有效促进MnO2溶解的可逆性,并在一定程度上提高电池的面积比容量(5.8 mAh·cm−2,静态条件)。
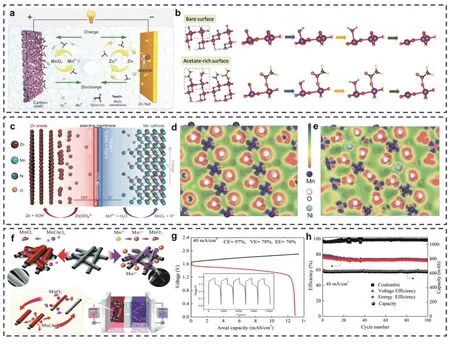
图8 (a)醋酸基电解质中Zn//MnO2电池示意图;(b) MnO2与裸露的表面和含有丰富的醋酸表面发生溶解反应的原子结构98。(c) Zn//MnO2解耦电池的充放电示意图以及(d)电极氧化的MnO2(101)和(e) Ni-MnO2(101)的电子密度差示意图106;(f) Mn(Ac)2(左上角)和MnSO4电解液(右上角)的电化学机制;(g)电解液为0.5 mol·L−1 Mn(Ac)2 +0.5 mol·L−1 Zn(Ac)2 + 2 mol·L−1 KCl的液流电池的充放电曲线以及(h)在40 mA·cm-2电流密度下的循环性能109Fig. 8 (a) Schematic diagram of Zn//MnO2 battery in acetate electrolyte. (b) Atomic structures for the dissolution reaction on MnO2 with a bare surface and with an acetate-rich surface 98. (c) Schematic illustration of Zn//MnO2 in the hybrid electrolytes during charge/discharge and electron density difference between (d) electrooxidized MnO2 (101) and(e) Ni-MnO2 (101) 106. (f) Electrochemical mechanism of Mn(AC)2 (top left corner) and MnSO4 electrolyte(top right corner). (g) The charge/discharge curve of the flow battery with electrolyte of 0.5 mol·L−1 Mn(AC)2 + 0.5 mol·L−1 Zn(AC)2 + 2 mol·L-1 KCl and (h) the cycle performance at current density of 40 mA·cm−2 109.
金属元素对电化学反应有催化作用,可促进活性锰的沉积作用,有效提高电池的反应动力学。Zhong等101报道了一种Co修饰的δ-MnO2正极材料,其中Mn(IV)和Co(III)的双催化作用促进了活性Mn的可逆沉积。其催化途径如下:
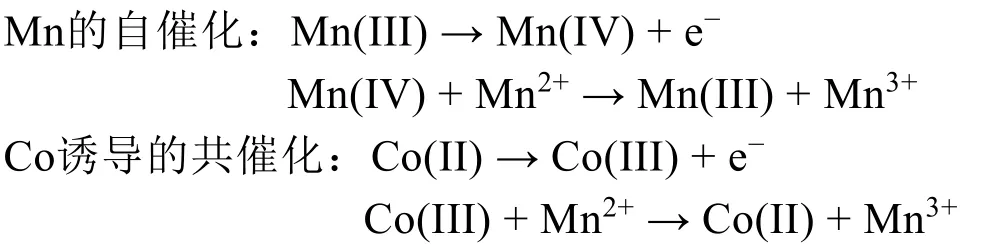
Co诱导的共催化有效促进了Mn的电化学沉积能力,因此,所制备的电池具有超过500 mAh·g−1的高比容量,且在经过5000次循环后仍保持63%的容量。
由于酸性电解液中存在大量质子,会促进析氢反应(HER),而析氧反应(OER)在碱性电解液中更容易发生102,103。在解耦电池体系中,离子交换膜能分离正/负极电解液,可同时实现正极电解液呈酸性,负极电解液呈碱性,这有效抑制了OER和HER的发生,从而扩大电化学窗口104。Zhong等105报道了一种电压平台高、反应速率快的Zn//MnO2解耦电池,负极的碱性电解液和正极的酸性电解液被一个中性室(K2SO4溶液)分隔,避免了酸性和碱性电解液的中和。得益于解耦电池独特的分离体系,其开路电压可达2.83 V,比能量密度为1621.7 Wh·kg−1。随后,Chao等106提出了一种将含Ni2+的电解液添加剂加入到解耦电池的正极电解液中的策略(图8c),利用强电负性金属Ni的催化作用(图8d和e),增强了活性电子态和电荷离域,即引入Ni会降低O的电子密度,电荷局部化作用减弱,改善了活性O 2p电子态,促进电荷转移,使得整个反应都在能垒较低的势能表面进行,进一步提高电池性能。这种Ni催化的解耦电池实现了高达50 mA·cm−2(50C,放电60 s,1C = 1 mA·cm−2)的反应动力学以及超高的电压平台2.44 V (1C)。此外,Huang等107提出了两性水系电解液解耦体系与锌锰电池的集成,HER和OER过程可分别在高电流密度下(高达1 A·cm−2)和低电流密度下进行,高/低电流密度可分别由高功率风能和低功率太阳能驱动,为未来可再生能源的存储/转换提供有效策略。需要注意的是,由于正/负极电解液的酸碱度不同,会不可避免地发生离子交叉,这在一定程度上降低倍率性能和能量密度。同时,昂贵的离子交换膜也会限制其大规模应用。
近年来,液流电池由于其高安全性、长循环寿命,以及能够完全释放发生结构崩塌时所产生的压力等优势,受到了广泛的关注108。Xie等109报道了一种高度可逆的中性Zn-Mn液流电池(图8f)。与MnSO4电解液相比,二氧化锰在含醋酸根(Ac−)的电解液中的氧化还原电位降低了530 mV,这是由于Ac−与Mn2+的配位作用造成的。在充电过程中,Mn(Ac)2转变成MnO2时会伴随着HAc生成,而HAc的吉布斯自由能比H+要小,驱动该反应所需的能量较少,从而降低了MnO2/Mn2+的电极电位。同时,Ac−的配位作用可使Mn2+直接转变成MnO2,而不会发生Mn3+的歧化,减少了副反应的发生,大大提高了循环稳定性。但由于电池库伦效率无法达到100%,放电时所产生的多余的游离H+会与Ac−结合产生醋酸,进而减弱Ac−的配位作用。而液流电池装置会不断更新电解液,稳定电解液中Ac−的浓度,在一定程度上保证了配位作用的实现。以0.5 mol·L−1Mn(Ac)2+ 0.5 mol·L−1Zn(Ac)2+ 2 mol·L−1KCl作为电解液制备的液流电池实现了优异的充放电性能(图8g)和循环性能(图8h)。此外,Li等110报道的一种无隔膜的液流Zn//MnO2电池,正极(Mn2+/MnO2)和负极(Zn2+/Zn)在同一种电解液中,无需使用离子交换膜,有效降低了成本。并且指出,需开发出一种具有良好的亲水性、优良的导电率和更大表面积的正极集流体,以实现较高的面积容量,为未来规模储能系统的发展奠定基础。
5 总结与观点
新一轮能源革命的核心为可再生能源发电与规模储能,在众多电化学储能技术中,水系锌二次电池由于其兼具经济性、安全性、生态友好以及高能量和高功率密度等优势而备受关注,成为储能领域的理想选择之一。电池的性能很大部分取决于电极材料,MnO2因其储量丰富、低成本、低毒性的优势,成为水系锌二次电池最佳候选正极。因此,Zn//MnO2电池的电极材料特性、储能机制以及针对容量低、循环寿命短等问题提出的优化措施被广泛研究。本文总结了当前主要的两类储能机理(嵌入-脱出和溶解-沉积机理)的特征及区别,以及针对这两种反应机制所提出的优化策略,为进一步推动具有高能量密度和长循环寿命的水系锌锰二次电池的开发提供研究方向。
5.1 嵌入-脱嵌机制与溶解-沉积机制的区别
近年来Zn//MnO2电池优异的电化学性能引起了广泛关注。目前主流的反应机理主要有嵌入-脱嵌机制和溶解-沉积机制两大类。嵌入-脱嵌机制主要在弱酸性的条件下发生。这种H+、Zn2+等离子嵌入-脱嵌行为会导致晶格间发生膨胀/收缩,并发生不可逆相变,生成副产物如ZnxMnO4、MnOOH、ZnMn3O7·3H2O和Zn2Mn3O8等,且活性材料还会溶解到电解液中,减小充放电容量。同时,Zn2+的高电荷密度会使Zn2+与MnO6晶格之间产生强静电反应,导致本征的离子扩散动力学较差,且由于反应机制和晶体结构的复杂性,难以确定Zn2+合适的调节位点。再加上嵌入-脱出机制的Mn4+/Mn3+氧化还原反应只利用了1个有效电子转移,限制了电池的容量和输出电压。这些问题大大影响锰基水系锌二次电池的发展。
近些年提出的溶解-沉积机制,是基于MnO2可逆溶解/沉积的行为实现电池的充放电,此过程与电解液的pH值有密切关系。该机制的电化学反应过程转移的有效电子数为2,其理论容量和理论电位均高于嵌入-脱出机制。Chao等62通过分析放电曲线中三个电化学窗口不同区域,解释了ZnSO4+MnSO4电解质中Zn//MnO2电池的储能机理。第一个区域(2–1.7 V)控制MnO2/Mn2+反应,第二个区域(1.7–1.4 V)和第三个区域(1.4–0.8 V)分别反映H+和Zn2+嵌入MnO2的结果。通过调控H2SO4的浓度,可控制D1高电压区的容量百分比,当H2SO4的浓度达到0.1 mol·L−1时,电池储能过程主要由溶解-沉积机制主导,实现了可观的放电电压和能量密度。然而,目前溶解-沉积机制复杂的内在反应路径尚未明晰,还无法实现锰基完全充分可逆的溶解-沉积,同时在酸性电解液中发生的锌腐蚀和析氢等问题会降低Zn沉积/剥离效率,这些问题仍待进一步研究。此外,一些报道曾提到在中性或弱酸性电解液中也能发生锰基可逆的溶解-沉积50,或在液流电池中也能观察到H+/Zn2+和MnO2共存现象110,说明溶解-沉积反应和H+/Zn2+的嵌入可能普遍存在于水系锌二次电池体系。
5.2 MnO2正极优化措施的特点
当前,蓬勃发展的储能市场使得对高性能电子设备和柔性可穿戴设备的需求愈发迫切,这极大推动了高能量密度和高安全的可充电电池的发展。本节总结了锰基电极优化措施的特点及其未来的发展方向。(1)针对嵌入-脱嵌机制:①采用涂覆涂层的方法,形成稳定SEI膜,抑制活性材料的溶解,或利用三维多孔网络结构为离子传输提供更多更稳定的通道,提高离子的嵌入-脱嵌速率;②与具有多孔结构的纳米材料进行复合,不仅能提高活性材料的比表面积,增大电极与电解液的接触面积,增加活性反应位点,从而提高电池的反应速率,还能避免添加剂的使用,实现更快的电子/离子传输速率;③材料在电池循环过程中会发生难以避免的内部结构崩塌现象,客体预嵌在扩大层状材料层间距的同时,还有助于稳定电极结构,并降低离子的扩散势垒,更有利于离子的嵌入/脱嵌;④离子参与氧化还原反应需要克服能量势垒,势垒越高,则发生反应时所需的能量越大,阻碍高功率、高能量密度电池的发展。异质元素具有与MnO2不同的电子构型,掺杂异质元素可诱导缺陷的形成,并调节MnO2的电子结构分布,从而减小离子间的静电斥力,降低电化学反应势垒,进而提高离子的嵌入/脱出速率。(2)针对溶解-沉积机制:通过调节电解液的组分或pH值,以降低能量势垒、促进锰基可逆溶解或提高未充分溶解的MnO2的活性的方法来调控MnO2的溶解-沉积行为。同时,金属元素对电化学反应的催化作用可有效提高二氧化锰溶解-沉积的可逆性。此外,解耦体系电池对正负极电解液的分离有效抑制了OER和HER,极大地提高了电池的电化学稳定性。同时,液流体系电池可通过消除浓度极化来维持电解液离子浓度的稳定,也能保证外来离子介质与Mn2+的协同作用的稳定实现,且流动的电解液在一定程度上抑制了锌枝晶的生成,进一步提高了电池的综合性能。除了对正极材料和电解液的研究,对于负极的析氢和腐蚀的影响也应该被考虑。
高性能水系锌二次电池在大规模电网储能领域具有广泛的发展前景,持续优化水系锌二次电池性能,进一步提高其能量密度、长循环稳定性、低成本及可回收利用性,推动其商业化进程。然而,目前Zn//MnO2电池领域仍存在如嵌入-脱嵌和溶解-沉积的内在机制未明晰,无法实现高能量密度等问题。如何有效结合两种机制,探讨高性能锰基电极的研发,制备出具有高能量密度和优异循环稳定性的实用水系锌二次电池,需要更加深入的研究。
猜你喜欢
中南民族大学学报(自然科学版)(2021年1期)2021-02-02
中学生数理化(高中版.高二数学)(2020年2期)2020-04-21
商品与质量(2019年42期)2020-01-17
山东冶金(2019年5期)2019-11-16
南方文学(2016年4期)2016-06-12
海峡科技与产业(2016年3期)2016-05-17
中国资源综合利用(2016年7期)2016-02-03
中国资源综合利用(2016年4期)2016-01-22
中国塑料(2015年11期)2015-10-14
中南大学学报(自然科学版)(2012年3期)2012-07-31
