
氟代溶剂在锂金属电池中的应用
2022-11-24 11:16:28何子旭陈亚威黄凡洋揭育林李新鹏曹瑞国焦淑红
物理化学学报 2022年11期
何子旭,陈亚威,黄凡洋,揭育林,李新鹏,曹瑞国,焦淑红
中国科学技术大学,中国科学院能量转换材料重点实验室,材料科学与工程系,合肥微尺度物质科学国家研究中心,合肥 230026
1 前言
自上世纪九十年代初锂离子电池(Lithium ion battery,LIB)发明以来,锂离子电池凭借能量密度高、循环寿命长、无记忆效应等优势,掀起了一场储能技术革命,逐渐占据了储能市场的主导地位,并且极大地推动了消费类电子产品和电动汽车等的应用和发展1,2。然而,近十年来,随着储能市场对于电池能量密度的需求不断增长,采用石墨作为负极的传统锂离子电池能量密度逐渐不能满足市场需求,有必要发展能量密度更高的储能电池体系3–5。相比于锂离子电池中的石墨负极,锂金属负极(Lithium metal anode,LMA)具有更高的理论比容量(~3860 mAh·g−1)和更低的还原电位(−3.04 V vs. SHE),有望能够大幅提升电池的能量密度,因此锂金属电池(Lithium metal battery,LMB)成为目前储能技术研究的前沿研究热点6。锂金属电池的正极可以采用传统锂离子电池的正极材料,也可以采用氧气或者单质硫等转化型正极材料。本文只讨论采用传统锂离子电池正极材料的锂金属电池,这类锂金属电池一般具有较高的工作电压,也称为高电压锂金属电池。
锂金属电池与锂离子电池的显著区别在于负极反应机制不同。锂离子电池采用石墨作为负极,在循环过程中Li+在石墨负极的层间进行插入/脱出反应,这种“摇椅式”电池结构本身具有较好的循环稳定性7,8。而锂金属电池采用金属锂作为负极,在充放电过程中,Li+在锂金属负极表面进行电化学沉积/剥离转化反应;由于金属锂具有极高的电化学活性,电解液在锂金属表面的副反应严重,并且在沉积过程中容易产生锂枝晶,导致电池内部短路和热失控等安全问题9–11。因此,发展锂金属电池面临一系列重大挑战:(1)锂金属负极在充放电过程中有巨大的体积变化,沉积过程中体积膨胀容易导致SEI (Solid electrolyte interphase)破裂,暴露的新鲜锂金属不断与电解液发生副反应,引发电池库仑效率(Coulombic efficiency,CE)降低、电解液干涸、电池寿命缩短等问题12;(2)金属锂不均匀沉积/剥离产生的锂枝晶可能刺穿隔膜造成内短路,引起电池燃烧甚至爆炸13;(3)锂枝晶容易从根部剥离,致使部分锂金属脱离与基底的连接形成“死锂”,“死锂”累积导致电池活性成分不断减少和电池界面阻抗的急剧增大14,15。
电解液是锂金属电池的核心,发展稳定的电解液体系,是解决锂金属电池问题的有效手段和策略。如图1a所示,电解液不仅起到传导锂离子的作用,同时电解液与负极/正极界面直接接触,由于大多数电解液在锂金属负极和高电压正极界面的热力学稳定性较差,会与电极在界面处发生副反应,从而在负极和正极界面分别形成SEI和CEI(正极电解质界面膜,Cathode electrolyte interphase),从而进一步影响电池的工作电压区间和循环稳定性16。电池的电化学窗口通常是由电解液溶剂化结构的最低未占据分子轨道(Lowest unoccupied molecular orbital,LUMO)和最高占据分子轨道(Highest occupied molecular orbital,HOMO)决定。理想情况下,电池工作电压区间在电解液LUMO和HOMO范围内,电解液作为一种化学惰性介质仅传输离子维持电荷平衡18。但是,对于锂金属电池而言,由于锂金属负极的化学势往往高于一般电解液的LUMO能级,因此电解液会在锂金属表面被还原;还原生成的固态副产物会形成SEI,SEI的组成和结构很大程度上决定了锂金属负极的稳定性和安全性。在正极一侧,高电压正极材料会导致电解液氧化分解,在正极界面形成CEI,其成分和结构对正极材料的结构稳定性发挥着关键作用19–21。由此可见,电解液的组分及其溶剂化结构决定了电解液与电极界面的反应性,进而影响了SEI和CEI的组成和结构,并最终决定了电池的电化学性能。因此,通过开发新型电解液体系或者新型功能添加剂的策略,能够有效调控电解液与正负极界面化学/电化学反应,优化CEI和SEI的组分和结构,从而保证锂金属电池能够安全、稳定、高效循环22–30。
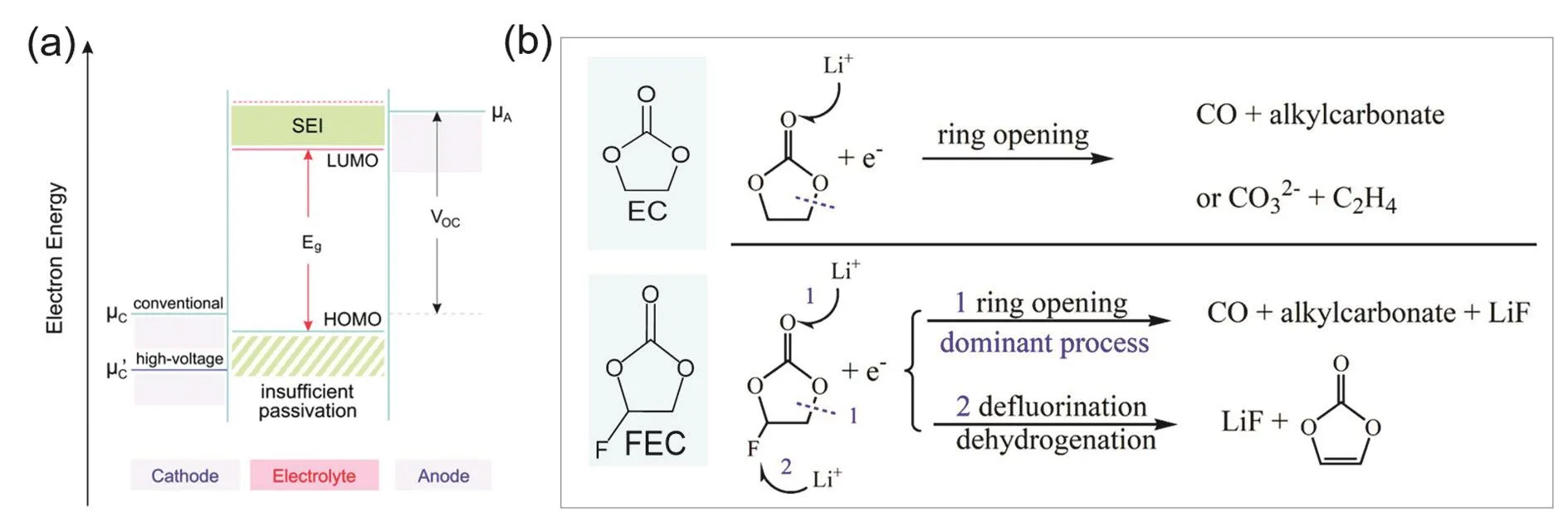
图1 (a)锂金属电池电极与电解液能级示意图16;(b) EC与FEC溶剂分解路径差异17Fig. 1 (a) Schematic diagram of energy levels of electrodes and electrolytes of LMB 16;(b) the decomposition path differences between EC and FEC solvents 17.
氟代溶剂在锂离子电池电解液中已经有广泛的应用和研究,目前氟代溶剂被证明能够有效改善电极界面稳定性、提升电解液的电压窗口。如图1b,使用氟原子取代碳酸乙烯酯(Ethylene carbonate,EC)中的一个氢原子,形成氟代碳酸乙烯酯(Fluoroethylene carbonate,FEC);由于溶剂分子结构和极性的变化,其在电极界面的分解路径会发生改变,得到成分和结构完全不同的SEI,进而影响电极材料的电化学性能31–33。文献研究表明,未经过氟取代的EC会在锂金属一侧分解产生较多乙烯(C2H4);而FEC分解产生的C2H4较少,且分解产物中含有较多氟化锂(LiF),后者在形成稳定的SEI中起到重要作用17。此外,将碳酸甲乙酯(Ethyl methyl carbonate,EMC)进行氟取代形成甲基三氟乙基碳酸酯(Methyl (2,2,2-trifluoroethyl)carbonate,FEMC)34,也可以有效提升溶剂的氧化稳定性,使得电池能够在更高的电压情况下安全运行35。
在锂金属电池中,氟代溶剂对电极界面性质的影响更大,因此对电池安全性和循环稳定性起到更为重要的作用。相比于锂离子电池,锂金属电池的界面稳定性更差,而氟代溶剂的加入在电极表面形成含有氟化锂的界面层(SEI和CEI),对提升锂金属电池的稳定性起到关键作用。最近,文献报道了多种基于氟代溶剂的锂金属电池电解液体系,有效提升了锂金属电池的循环性能,推动了锂金属电池的发展。例如,局域超浓电解液中的稀释剂多为高度氟代的溶剂分子36,对常用溶剂进行结构设计形成部分氟代溶剂23,将特定氟代官能团引入溶剂分子中37等,这些策略都在不同程度上改善了锂金属电池的循环性能,提升了电池的安全性。本文综述了近年来应用于锂金属电池中的主要新型氟代溶剂,并分析了其在电解液中的作用,以及与电池性能之间的构效关系;进而总结了构建氟代溶剂分子的策略和思路,期望能对电解液溶剂分子的设计与开发起到一定的借鉴启发作用。
2 氟代溶剂作为电解液稀释剂
2.1 超浓电解液与局域超浓电解液
基于电解液的电导率、粘度、成本等多方面考虑,锂电池的电解液浓度一般选择在~1 mol·L−1左右,其综合性能最优。在这种常用的低浓度电解液中,大部分溶质被完全解离,因而Li+的第一溶剂化鞘层中仅存在溶剂分子,形成溶剂分离离子对结构(Solvent-separated ion pair,SSIP,图2a);并且电解液中存在着大量游离的自由溶剂分子和阴离子。当进一步提升电解液中锂盐的浓度,形成超浓电解液(High concentration electrolyte,HCE)体系,这类电解液的溶剂化结构与低浓度电解液有较大的差别。在超浓电解液中,溶剂分子数量的急剧下降,造成大量溶质不能完全解离,形成接触离子对结构(Contact ion pair,CIP,图2b)以及多个溶质和溶剂分子共同作用形成的聚集体结构(Aggregate,AGG,图2b)38,39。
超浓电解液以其独特的溶剂化结构表现出与低浓度电解液完全不同的性质,因而相较于后者具有诸多优点:(1)溶剂化结构的改变降低了阴离子的LUMO能级,同时Li+会倾向携带更多阴离子到达负极表面,因而阴离子在锂金属表面优先被还原,形成阴离子分解主导的SEI,对锂金属负极具有更好的保护作用41–43;(2)溶剂分子的减少降低了Li+的屏蔽作用,大幅提升了Li+迁移数;(3)几乎所有的溶剂分子都将孤对电子部分给出参与配位,游离的自由溶剂分子减少,因此提升了电解液的热稳定性,增强了电解液氧化稳定性44–46(图2d)。
然而,超浓电解液仍存在较多问题,尤其是其粘度较大且锂离子电导率较低,严重影响了电池的倍率性能和工作温度区间。随着浓度的增加,溶质与溶剂之间的相互作用增强,电解液粘度增加、润湿性降低,电解液存在难以浸润隔膜、电解液动力学变差等问题47。另外,从经济成本的角度来看,增加电解液中盐的比例会导致成本上升,不适于大规模生产48。针对上述问题,Zhang团队49在2017年首次提出通过向超浓电解液中加入一种高度氟代的链状醚,1,1,2,2-四氟乙基-2,2,3,3-四氟丙基醚(TTE),在较好地保持超浓电解液溶剂化结构的同时,解决了超浓电解液粘度大、电导率低的问题。如图2c所示,氟代醚的加入,分割了超浓电解液的网络结构,形成局部聚集的溶剂化结构,即局域超浓电解液(Localized high concentration electrolyte,LHCE)。自此局域超浓电解液成为锂金属电池的研究热点,近年来有大量相关文献报道该类型电解液用于锂金属电池的研究。
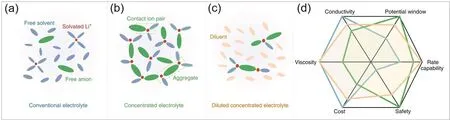
图2 (a)传统电解液、(b)超浓电解液、(c)局域超浓电解液溶剂化结构示意图;(d)三种电解液性能比较40Fig. 2 (a–c) Solvation structure diagram and (d) performance comparison of conventional electrolyte, HCE and LHCE 40.
为了获得电化学性能和理化性质俱佳的电解液体系,对稀释剂的选择有一定的要求。Yamada等40提出,稀释剂需要具备以下特征:(1)低粘度,能够显著降低超浓电解液的粘度;(2)成本低,能够降低电解液的总成本;(3)适当的介电常数和配位能力,使超浓电解液具有高溶解度,同时不改变超浓电解液的局部配位环境;(4)足够的惰性/稳定性,不影响超浓电解液的电化学窗口;(5)不易燃且挥发性低,能够保证超浓电解液的安全性。氟代溶剂是目前报道最多的稀释剂,氟元素的电负性在所有元素中最大,并且其原子半径较小、空间位阻较低。通过对溶剂分子进行氟取代或者多氟取代,氟原子强烈的吸电子作用使得溶剂分子难以给出电子进行配位而成为一种惰性溶剂。因此,氟代溶剂能够与其他溶剂互溶但并不解离溶质分子,也不改变电解液的局部配位环境。同时,氟代溶剂自身的惰性使得其具有粘度低、稳定性高等优点,很好地满足了稀释剂的要求。近年来开发的局域超浓电解液与稀释剂的结构如图3与表1所示。
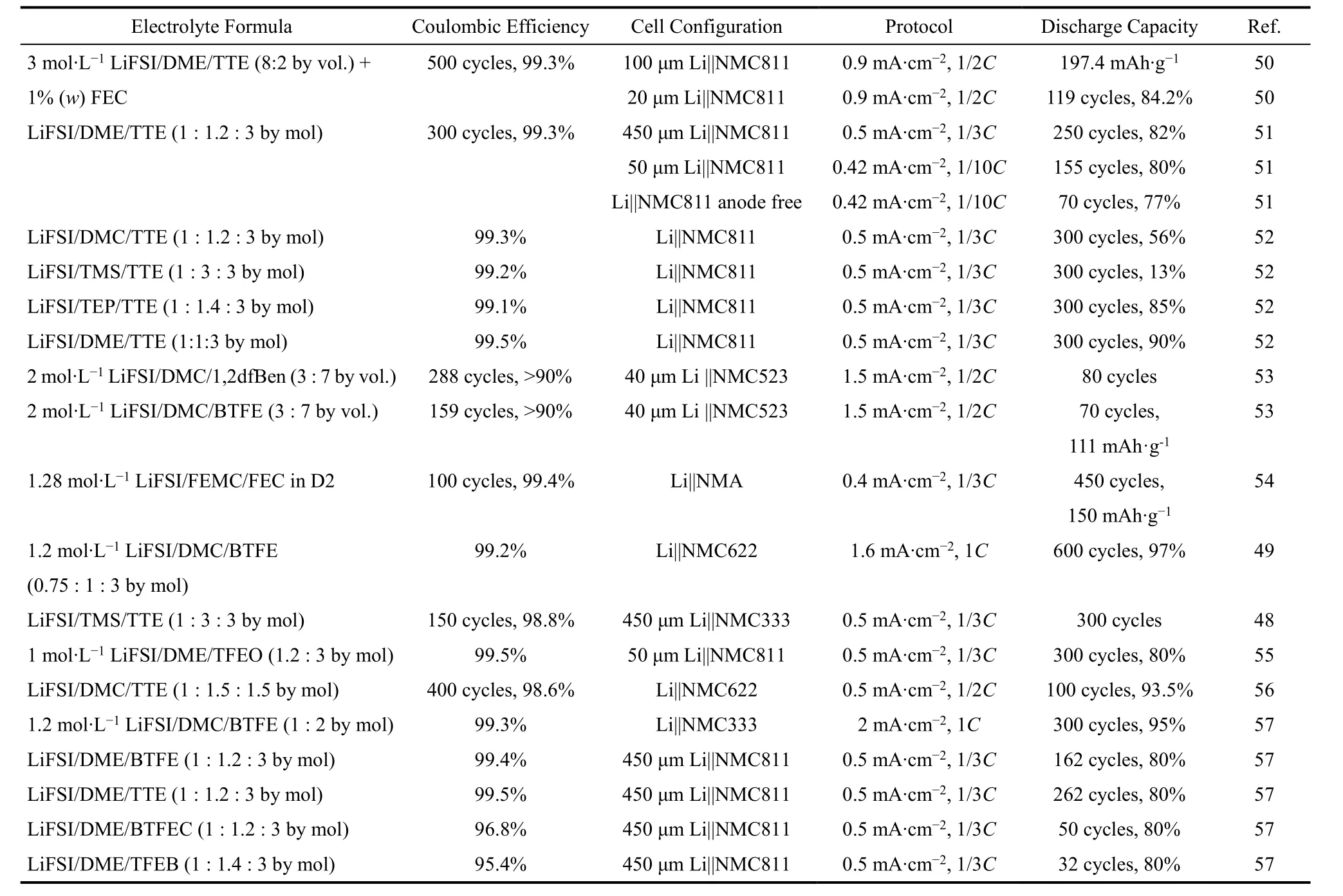
表1 局域超浓电解液配方与性能Table 1 Localized high-concentration electrolytes formula and the battery performance.
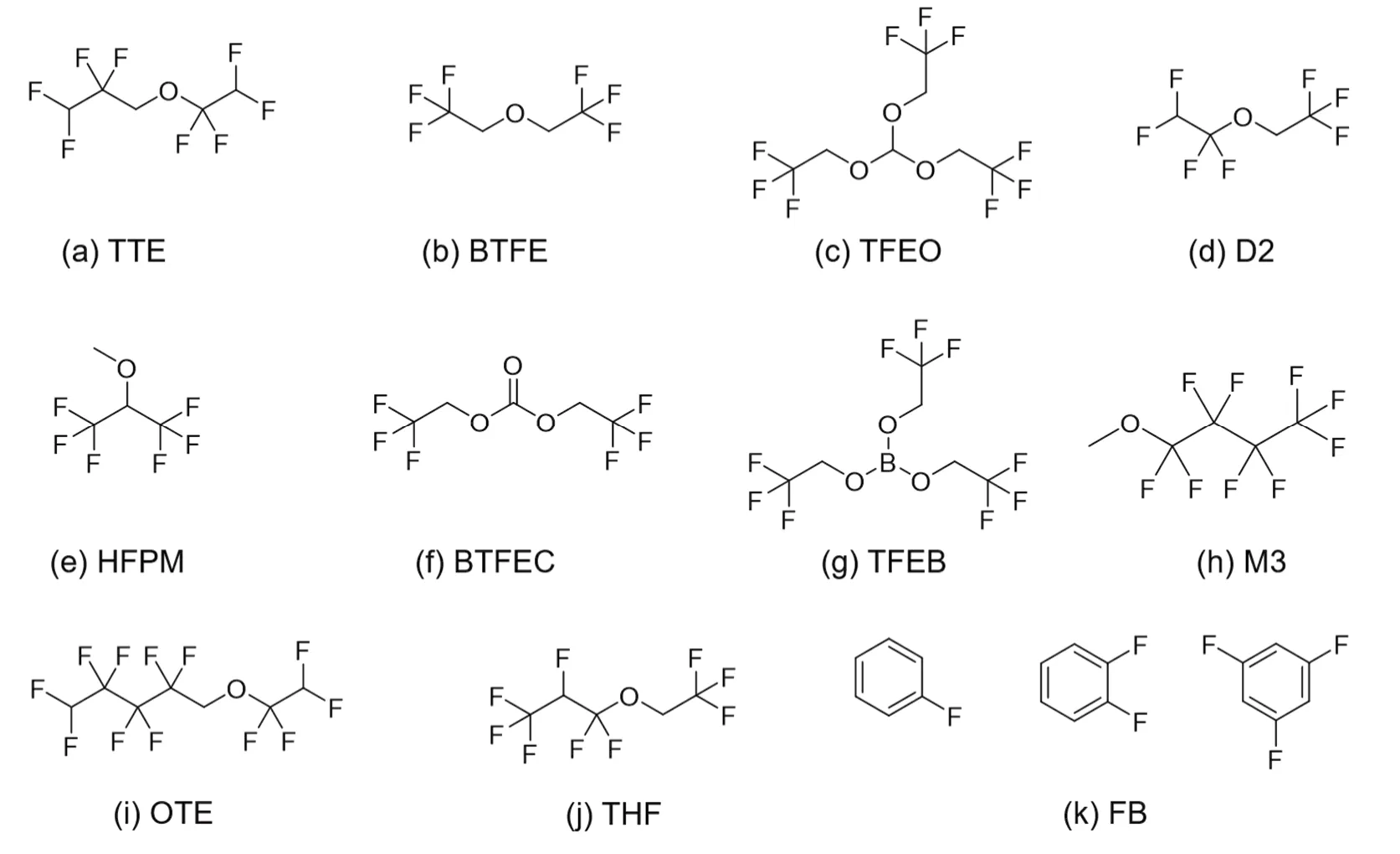
图3 稀释剂分子结构示意图Fig. 3 Schematic diagram of diluent molecules.
2.2 稀释剂的作用
稀释剂作为一种高度氟代溶剂,其溶剂分子结构上F取代的位点较多,并且距离官能团位置较近,F强烈的吸电子能力使得官能团难以给出电子与Li+配位,导致难以溶解Li盐。在电解液中加入稀释剂,不会改变原有的溶剂化结构,稀释剂更多地出现在溶剂化鞘层的外围,在调制电解液理化性质的同时参与SEI和CEI的构建。因此,稀释剂不仅作为惰性溶剂起到“稀释”的作用,还对电解液的理化性质、电解液溶剂化结构、电池正负极界面性质有重要影响。
2.2.1 对电解液理化性质的影响
Piao等56研究了不同比例的LiFSI/DMC/TTE电解液(LiFSI:双氟磺酰亚胺锂;DMC:碳酸二甲酯)的理化性质与电池性能,提出稀释剂是一种反溶剂(在工业中可以通过向溶液中加入与溶剂互溶而不能溶解溶质的反溶剂来析出溶质)。图4a为不同比例LiFSI/DMC/TTE电解液的一些理化性质;从图中可以看出随着稀释剂比例的上升,溶液逐渐饱和,甚至出现浑浊现象。稀释剂的加入可以显著降低电解液的粘度,维持电解液的电导率。对电解液与隔膜(Celgard 3501)之间的接触角测试发现:1 mol·L−1LiPF6/EC/DMC、LiFSI/DMC (摩尔比1 : 1.5)、LiFSI/DMC/TTE (摩尔比1 : 1.5 : 1.5),以上三种电解液的接触角分别为51.8°、62.4°、32.5°,说明稀释剂的加入改善了电解液对隔膜的润湿能力。同时,燃烧测试也表明LiFSI/DMC/TTE电解液具有良好的阻燃性。
Yao等58利用分子动力学模拟对超浓电解液和局域超浓电解液的介电常数进行了研究,发现锂盐浓度的增加使得电解液中具有CIP溶剂化结构的比例上升,并且提升了介电常数;由于相应的自由溶剂比例下降会降低电解液的介电常数,因而介电常数对于锂盐浓度的曲线呈现出火山图状。而对于LiFSI/DMC/TTE和LiFSI/DME/TTE (DME:乙二醇二甲醚)体系,随着稀释剂TTE比例从0%增加到71.4%,电解液的介电常数呈线性增长趋势(图4b)。其原因在于稀释剂的介电常数大于溶剂分子的介电常数,并且稀释剂不参与配位,因此加入较多的稀释剂会将超浓电解液体系的三维网络结构分割开,因而在稀释剂比例较高(> 50%)时,曲线变得平缓。Ding等59进一步将稀释剂的介电常数以及稀释剂与Li+的结合能作为评价稀释剂优劣的参数,该研究发现:与DME相比,具有更低介电常数和更大结合能的稀释剂能有效地增强Li+与阴离子的结合强度,从而导致电解液中更多聚集体结构(AGG)的出现,有利于阴离子的完全分解(图4c)。介电常数代表电介质对外界电场的反应,而电导率代表电解液中电荷流动的难易程度。在局域超浓电解液中,随着稀释剂的比例上升,电解液中传导电荷的离子数减少,电导率会有一定程度的下降。
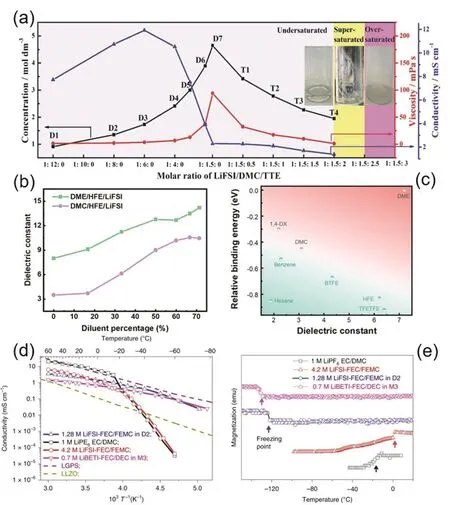
图4 (a)不同比例LiFSI/DMC/TTE电解液理化性质56;(b)介电常数随稀释剂比例的变化曲线58;(c)不同溶剂与Li+的结合能和介电常数59;(d,e)电解液电导率与磁化率随温度变化曲线54Fig. 4 (a) Physicochemical properties of LiFSI/DMC/TTE in different proportions 56; (b) curves of dielectric constant as a function of diluent ratio 58; (c) binding energy and dielectric constant of different solvents with Li+ 59;(d, e) conductivity and magnetic susceptibility of electrolyte vary with temperature 54.
普通电解液的熔点较高、去溶剂化能较高,致使电池低温性能衰减严重,难以在低温下工作60。但稀释剂极化能力弱、分子间相互作用力较小,因此熔沸点一般较低。将稀释剂引入到电解液体系中,可以降低电解液的熔点,使得电解液能够在低温条件下稳定运行。如图4d–e,Fan等54开发了两种 局 域 超 浓 低 温 电 解 液 : 1.28 mol·L−1LiFSIFEC/FEMC/D2 (D2:1,1,2,2-四氟乙基-2,2,2-三氟乙 基 醚 ) 和 0.7 mol·L−1LiBETI-FEC/DEC/M3(LiBETI:双(五氟乙基磺酰基)亚氨基锂;DEC:碳酸二乙酯;M3:甲基九氟丁醚)。以上两种电解液的电导率随温度降低而缓慢降低并维持在较高水平,但普通电解液则出现明显拐点(当温度低于一定值后电导率迅速降低)61。通过测量电解液的磁化率随温度的变化曲线发现,两种电解液的熔点均低于−120 °C,这也是两者在低温下仍能保持较高电导率的主要原因。
2.2.2 稀释剂对溶剂化结构的调控
有些稀释剂分子由于氧原子的存在,具有一定的锂离子配位能力,因此对电解液的溶剂化结构具有一定的调节作用。通过拉曼光谱,可以测量分子的振动、转动能级,从而获得电解液体系的结构信息。在电解液中,SSIP、CIP、AGG结构的溶剂化结构不同,分子间相互作用力存在差异,因而拉曼峰值会有稍许偏移。如图5a–b,Cao等30在对LiFSI/DME/TFEO (TFEO:三(三氟乙氧基)甲烷)体系中稀释剂TFEO的含量进行调控时发现,随着电解液中TFEO比例上升,在拉曼光谱中几乎没有出现游离DME溶剂的峰,并且电解液中AGG结构的含量略有上升。Ding等59在LiFSI/DME/D2体系的研究中通过对拉曼光谱中FSI−的峰值分析也得出了相似结论:在加入稀释剂后,阴阳离子之间的相互作用更加强烈,FSI−主要存在于AGG结构中。但是Ren等62的研究(图5c)发现,在LiPF6/DMC/HFPM(HFPM:1,1,1,3,3,3-六氟异丙基甲基醚)体系中,随着稀释剂比例的上升,拉曼光谱数据中出现了自由溶剂的峰,同时CIP结构比例增加、AGG结构比例减少;并且将稀释剂替换为TTE或者在LiFSI/DME/TTE体系中都有相同趋势。作者认为这是由于稀释剂与溶剂之间的偶极与偶极相互作用,将AGG结构分割成了CIP结构,并且出现了完全由稀释剂包围的自由溶剂分子。
2019年,Ren等51开发了LiFSI/DME/TTE体系,并深入研究了该电解液体系的溶剂化结构。通过对电解液进行了NMR-17O测试发现,随着TTE稀释剂的加入,DME中氧原子的化学位移减小,而阴离子FSI−和稀释剂TTE中氧原子的化学位移无明显变化。该研究结果证明TTE稀释剂不会干扰FSI−和Li+在高浓电解液中的强相互作用,而且会使更多的FSI−阴离子进入内溶剂化鞘层。LiFSI/DME/TFEO体系30也有相同现象,稀释剂分割大团簇,并与DME之间存在相互作用,从而引起DME化学位移的改变(图5b)。Amanchukwu等63进行NMR-7Li测试也发现加入TTE稀释剂会使得Li+的化学位移减小,从另一方面验证了稀释剂能够增强阴阳离子间的相互作用。
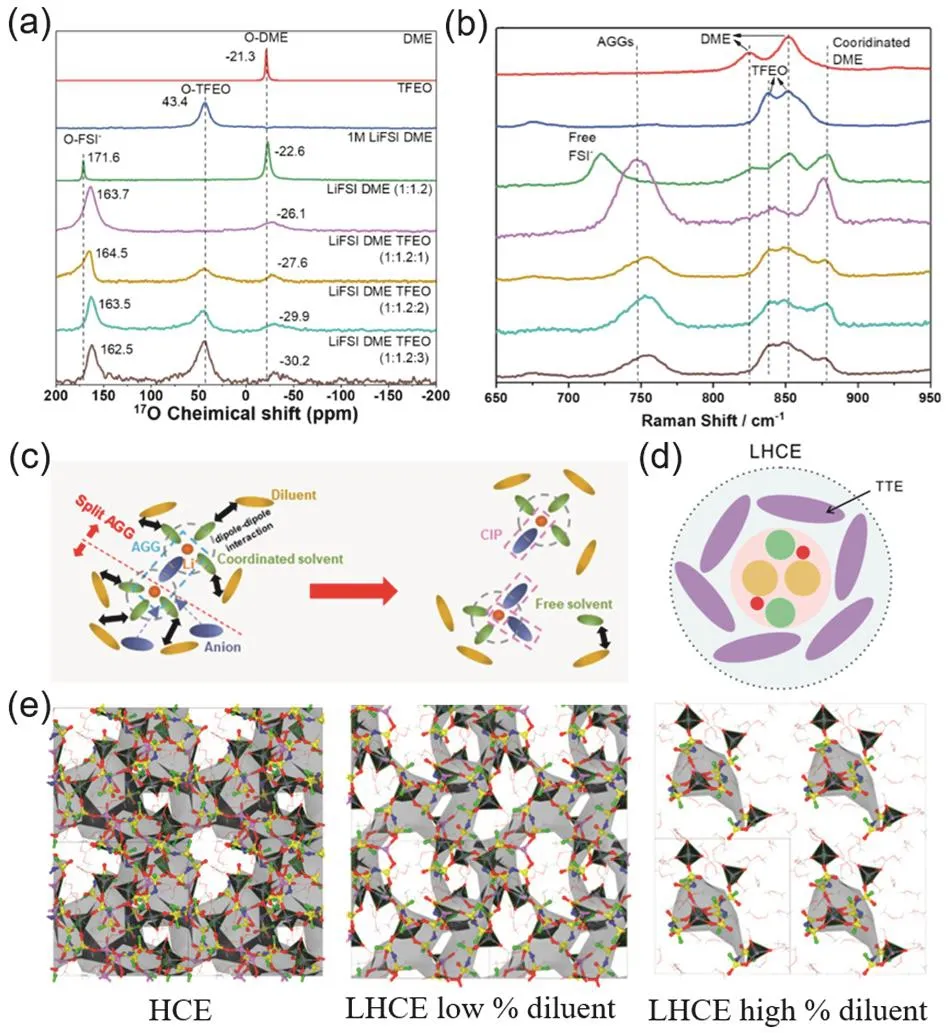
图5 (a,b)不同比例LiFSI/DME/TFEO电解液的拉曼光谱和NMR-17O数据30;(c)稀释剂将AGG结构分割为CIP结构示意图62;(d) LHCE溶剂化结构示意图51;(e)理论模拟HCE、LHCE电解液结构64Fig. 5 (a, b) Raman spectra and NMR-17O data of LiFSI/DME /TFEO electrolyte in different proportions 30;(c) thinner divides the AGG structure into CIP schematic 62; (d) schematic diagram of solvation structure of LHCE 51; (e) structure of HCE and LHCE electrolyte under theoretical simulation 64.
实验中对电解液进行拉曼光谱、核磁共振谱的分析能够提供电解液的整体信息,实验数据表明稀释剂存在于溶剂化鞘层周围但不参与溶剂化结构,对溶剂化层提供保护和屏蔽作用(图5d)51。如图5e,Zhang团队64对局域超浓电解液LiFSI/DMC/BTFE (BTFE:2,2,2-三氟乙醚)49进行理论计算模拟,发现当电解液体系中稀释剂比例不高(LiFSI : DMC : BTFE = 0.94 : 1.10 : 0.65,摩尔比)时,由阴离子FSI−主导的三维结构网络不会被破坏而是被稀释剂撑开;但当稀释剂增加到一定比例(LiFSI : DMC : BTFE = 0.64 : 1.10 : 1.65,摩尔比),三维网络结构会被分割成岛状溶剂化结构,部分Li+保持超浓电解液中的AGG结构,但部分Li+仅与一个FSI−配位,甚至出现Li+与稀释剂分子BTFE配位的现象。该现象能够解释加入稀释剂后电解质的溶解度降低,说明稀释剂与Li+、溶剂分子、阴离子之间都存在相互作用。该团队对LIFSI/DME/TTE体系的分子模拟研究发现65,TTE无法通过O原子但可以通过F原子与Li+发生相互作用而参与配位,但TTE分子与Li+的相互作用力相较于阴离子或溶剂较弱,因此容易从溶剂化鞘层中脱出。在稀释程度较高的局域超浓电解液中,两个岛状三维网络之间无溶剂分子,也几乎无Li+。该研究认为Li+可以通过短暂地与稀释剂TTE中F原子作用,形成由TTE包围的溶剂化结构;但随后TTE迅速脱去,Li+进入到另一个岛状网络结构中,该现象类似于Li+在电解液的两个岛状网络结构中进行跳跃,从而实现Li+的传输。
2.2.3 稀释剂对正负极界面的影响
超浓电解液中阴离子与Li+发生相互作用,使得LUMO能级降低,因而产生阴离子分解主导的SEI。XPS (X射线光电子能谱)等实验数据表明:阴离子分解主导的SEI中含有大量的LiF等无机成分并且分布均匀,导致SEI具有较高的机械强度和较大的表面能;因而在SEM、TEM的形貌观察中,超浓电解液中锂金属的沉积形貌呈现大块状并且十分平整41。由于稀释剂是高度氟代的溶剂分子,具有强烈的吸电子能力,因此其LUMO能级较低,容易与锂金属负极发生副反应。
Lee等50对LiFSI/DME/TTE电解液体系的研究发现:LiFSI/DME/TTE电解液的首圈库仑效率仅有40%,但却表现出优异的循环稳定性和较长的电池寿命。实验中验证在首圈锂沉积的过程中,TTE分解消耗大量Li+和电子而形成稳定均匀的SEI。通过飞行时间-二次离子质谱(TOF-SIMS)进一步证明在没有电场的条件下,TTE也能够在锂金属表面迅速形成一层富含LiF的界面层。如图6a,对LiFSI/DME和LiFSI/DME/TTE两组的XPS数据进行对比分析发现:LiFSI/DME/TTE体系中出现了独有的C-F和C-F2峰,也证明了TTE会参与SEI形成过程,此现象在Dong等66的研究中也被证实。
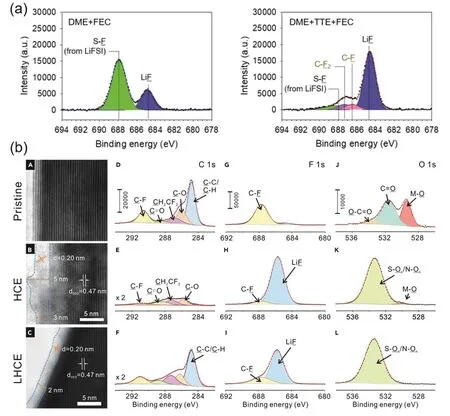
图6 (a) XPS证实稀释剂参与SEI构建50;(b) Li||NMC811在LiFSI/DME与LiFSI/DME/TTE中循环50圈后正极界面TEM图与XPS 51Fig. 6 (a) Diluents involved in SEI construction confirmed by XPS 50; (b) TEM image and XPS of the Li||NMC811 in LiFSI/DME and LiFSI/DME/TTE after 50 cycles 51.
Zheng等67运用理论计算详细模拟分析了LiFSI/DME/TFEO体系中电解液的分解过程,并给出了分解反应的机理与步骤。该研究推测锂盐LiFSI通过四电子分解机理,早期产生含有F和N(SO2)2等基团的产物,并主导着SEI的成分。稀释剂TFEO的分解一般需要六个电子,比LiFSI更为稳定,但也会参与构建SEI。该研究还阐明了在阴离子攻击下TFEO脱氢反应的机理,说明稀释剂与阴离子间存在相互作用。值得注意的是,溶剂分子DME在整个过程中都十分稳定,并没有发生化学反应的倾向。
如图6b,Ren等51对在LiFSI/DME/TTE体系中循环50圈后的NMC811正极表面进行研究发现:相比于超浓电解液中形成的CEI,局域超浓电解液中形成的CEI更薄(2 nm)且更均匀。从XPS数据也可以看出局域超浓电解液中正极表面的C―H、C―C键的成分明显增加,并且有更多的C−F信号,推测稀释剂在正极表面发生分解,更好地保护了正极界面。
Cao等57对于电解液中稀释剂的作用有十分系统的研究。该研究以LiFSI/DME体系为基础电解液,选用五种稀释剂BTFE、TTE、BTFEC (二(2,2,2-三氟乙基)碳酸酯)、TFEB (三(2,2,2-三氟乙基)硼酸酯)、TFEO。实验研究表明:稀释剂不仅可以保持高浓电解液体系的溶剂化结构,对界面钝化层也有显著影响,围绕在溶剂化鞘层周围的稀释剂可以通过共同参与SEI的形成,从而加速或者减缓阴离子的分解。由于BTFEC是氟代碳酸酯,其碳氧双键的极化能力较强,仍具有一定溶解盐的能力,因而在理论计算中出现在溶剂化鞘层中;但是BEFEC参与溶剂化结构破坏了原有电解液中阴阳离子部分解离的结构,造成电解液性能大幅下降和锂金属沉积形貌不均匀,是一种不良稀释剂。由此可知,不同的稀释剂与锂金属的作用存在差异,在TTE-LHCE中形成的SEI富含Li2O,且厚度分布最为均匀,稳定锂金属表面的效果最好。除此之外,不同稀释剂对正极也有不同的影响。在TFEBLHCE的SEI中,可以检测到Ni元素的存在,说明在该电解液中正极结构中的Ni溶出较为严重。结合对正极的XPS和TEM等数据的分析,发现TFEB显著加速了富镍NMC811材料的氧释放,并且由于B元素的缺电子性质,导致了正极的快速衰变;而BTFE、TTE和TFEO表现十分良好,在正极界面形成了薄且均匀的CEI,因而基于BTFE、TTE和TFEO的LHCE分别表现出99.4%,99.5%和99.5%的高库仑效率。Li||NMC811全电池的容量保持率顺序为TFEO-LHCE > TTE-LHCE > BTFE-LHCE >BTFEC-LHCE > TFEB-LHCE。Ren等52在LiFSI/DME/TTE体系中,替换多种不同溶剂进行比较,从另一个角度阐述了稀释剂与阴离子、溶剂会发生协同作用,参与SEI与CEI的构建。
上文所提及的稀释剂大多数为氟代醚,而苯及其衍生物由于低极性、高对称性等优点也可以作为稀释剂。Xie团队68,69将苯进行氟取代合成氟苯(FB)、Yoo等53对苯进行双位点氟取代成为邻二氟苯等,成为除氟代醚外的重要稀释剂。氟代苯容易制备且经济成本低,且对苯进行氟取代可以进一步提升苯的抗氧化性。同时,氟代苯使得其对称性被破坏,对苯分子的共轭键有一定影响,因此其C―F键比一般的C―F键更加活泼。通过能量色散X射线光谱(EDS)元素分析发现,在氟苯中浸泡的锂金属表面出现了氟元素,并且该电解液在循环中产生的SEI也富含LiF。Yoo等在文中也提及除邻二氟苯以外,间二氟苯、对二氟苯、甲苯等都能够提升LiFSI/DMC体系的电池性能。
综上所述,通过精细的分子结构设计F原子取代的位置与数量,或者通过调控溶剂与稀释剂的比例等手段,都可以进一步优化含有稀释剂的电解液,从而提升锂金属电池的循环稳定性和安全性。但稀释剂在锂金属电池中的应用也存在一些问题:(1)加入稀释剂存在反溶剂效应而导致电解液溶解盐的能力下降,不利于电解液的储存;(2)稀释剂对参与构建SEI和CEI界面层的影响目前没有明确机理,作用存疑;(3)大多数稀释剂成本较高,需要大量氟化物进行合成,现有的稀释剂种类较少。
3 氟代醚作为电解液的主溶剂
超浓电解液最大的亮点在于其独特的溶剂化结构,即提升电解液中CIP、AGG分子团簇的比例,构建离子的三维网络。近年来,发展了诸多新方法构建类似的溶剂化结构,例如向溶液中加入金属有机框架材料(MOF等),构建完全由CIP结构组成的电解液70,71;向局域超浓电解液中加入特殊的添加剂,可以稳定AGG结构72等。超浓电解液中AGG结构形成的本质是由于溶剂量较少,Li+与阴离子无法被完全解离。因此,选用弱溶剂化能力的溶剂具有一定溶解盐的能力,但不至于使锂盐完全解离,例如乙醚73、1,4-二氧六环74等,也能够构建具有离子通道的三维网络。
增加碳链长度可以增加电解液的稳定性,并维持电解液溶解锂盐和传导锂离子的能力。对于醚类溶剂分子来说,需要由其分子中O原子上的孤对电子与锂离子进行配位,从而溶解锂盐。通过对醚类溶剂分子进行选择性氟代,可以调节醚类溶剂分子的溶剂化能力,从而构建不同类型的溶剂化结构。值得注意的是,由于F原子的强吸电子能力,仅有和O原子有一定距离的位置上(β-C)进行部分F取代,才能维持电解液溶解盐的能力。Bao和Cui团队将常见醚类溶剂的碳链增长并部分氟代,在这方面做了大量的建设性工作,开创了电解液设计的新领域。其所用主要配方与溶剂结构见表2与图7。
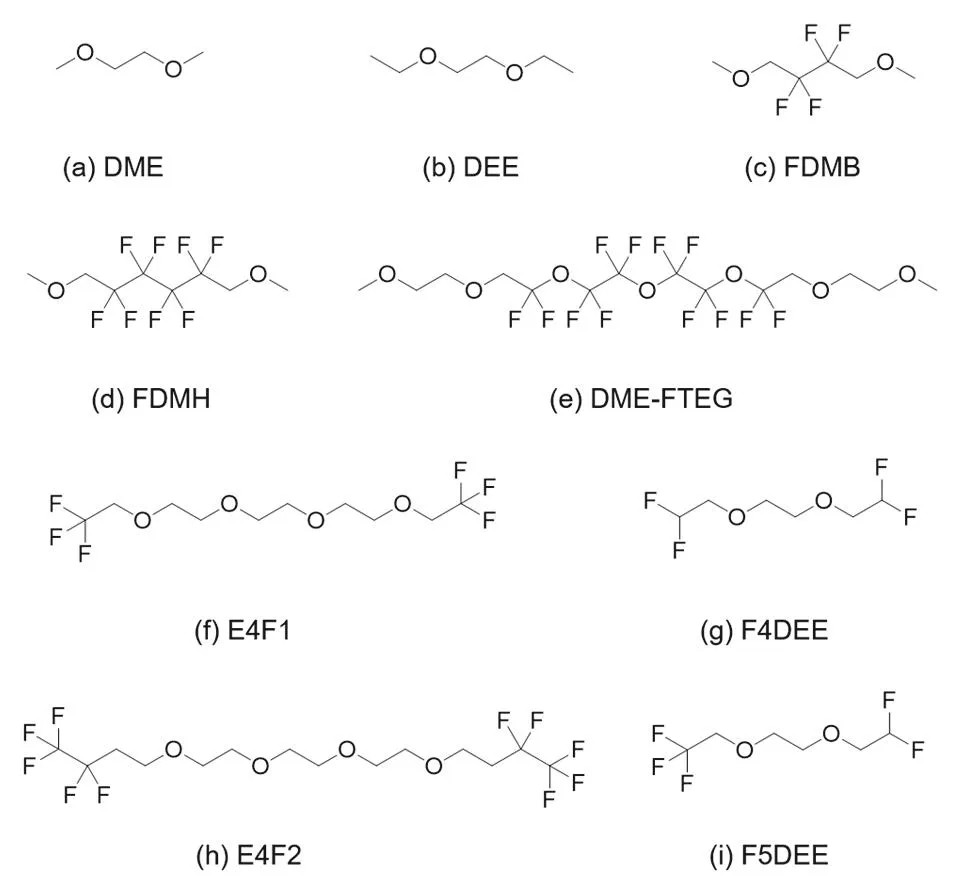
图7 部分氟代溶剂分子结构Fig. 7 Molecular structure of partially fluorinated solvents.
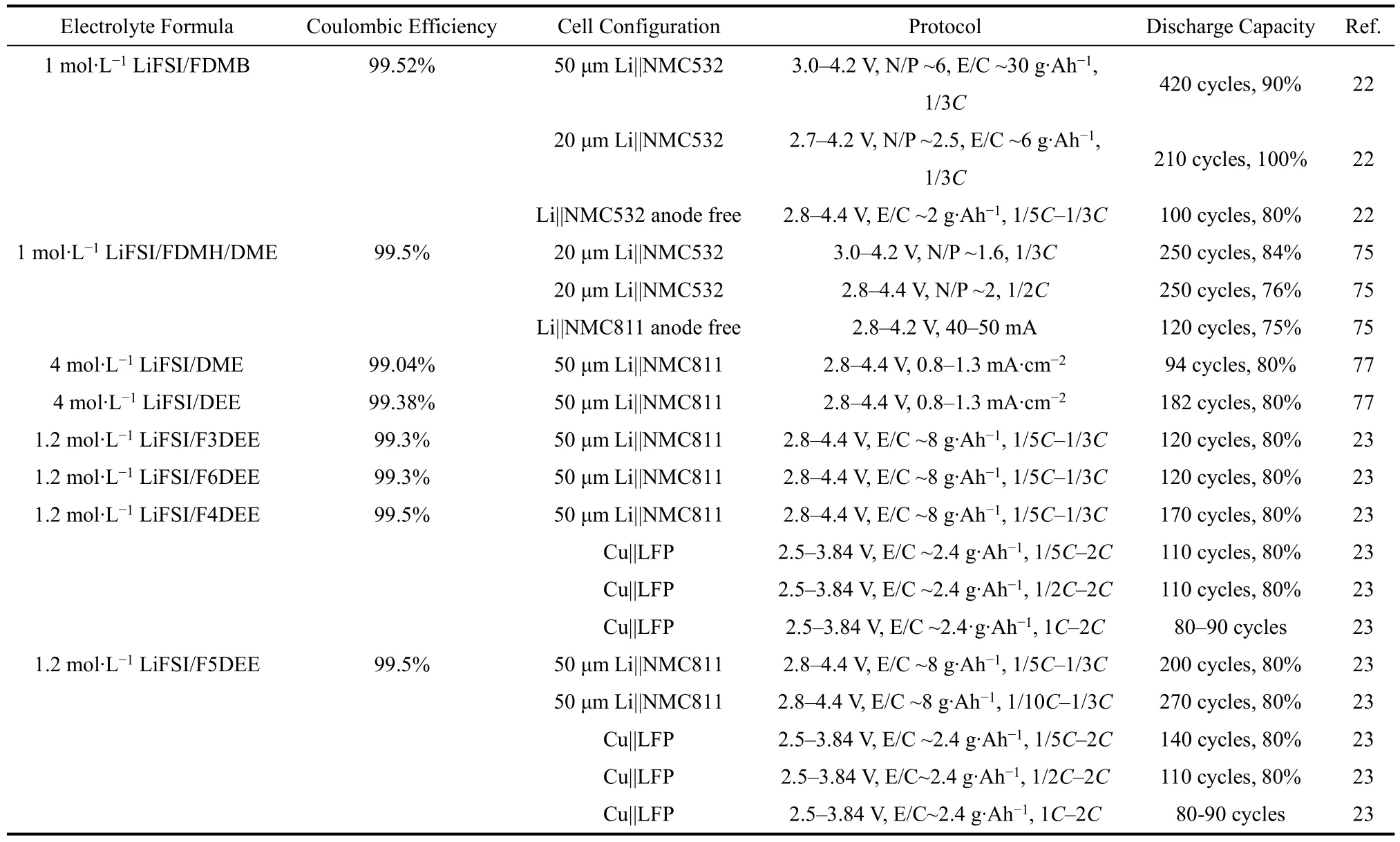
表2 部分氟代溶剂电解液配方及其电池性能Table. 2 Partially fluorinated solvent electrolytes formula and battery performance.
3.1 中间碳链增长并部分氟取代
2020年,Bao和Cui团队22设计了氟代醚FDMB分子(图7c)用于锂金属电池电解液中。该分子设计中将DME分子的中间碳链增长,并在新增的C原子位置上进行F取代,得到了FDMB分子。1 mol·L−1LiFSI/FDMB电解液中,锂金属负极的首圈库仑效率高达~97.6 %,随后仅在5个充放电循环内库仑效率快速上升至> 99%,并且该电解液能够很好与正极匹配。该研究通过分子模拟、核磁、紫外-可见光光谱等手段分析了溶剂化结构,发现FDMB中的―O―和β-C上的F可以共同作用于Li+,构成一个五元环,因此电解液呈现特有的棕色(FDMB分子本身呈无色)。但F原子与Li+的作用相较于DME中两个―O―的螯合作用较弱,降低了Li+的溶解能力。如图8所示,在1 mol·L−1LiFSI/DME体系中,出现在Li+周围的阴离子和溶剂比例为FSI−: DME =2.31 : 1,而在1 mol·L−1LiFSI/FDMB体系中该比例为3.29 : 1,即FDMB分子使得更多的FSI−出现在内溶剂化鞘层。该电解液类似于局域超浓电解液,产生阴离子主导的SEI。而对Li沉积形貌和SEI的分析发现FDMB体系产生的SEI主要成分中含有F、S、O等元素,SEI厚度均匀而且非常薄。该研究证明1 mol·L−1LiFSI/FDMB中形成的SEI中更多的由阴离子分解产生,并且有更强的自限制能力,能更好地保护锂金属。但是在LiFSI/FDMB体系中,锂金属的沉积过电势稍有增加,这可能与氟化导致的离子电导率下降和高SEI阻抗相关,也可能是由于锂沉积形貌表面积较小造成的。
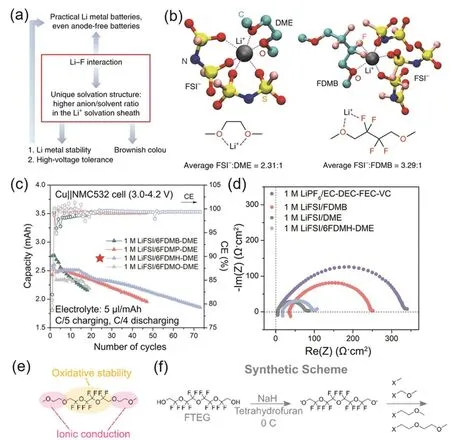
图8 (a) FDMB中Li-F作用示意图22;(b) LiFSI/DME和LiFSI/FDMB体系Li+配位情况的理论优化结果22;(c,d) LiFSI/6FDMH-DME系列电解液性能与阻抗75;(e) DME-FTEG分子构效关系76;(f) DME-FTEG系列分子合成方法76Fig. 8 (a) Diagram of Li-F interaction in FDMB 22; (b) theoretically optimized structures of ligands of LiFSI/DME and LiFSI/FDMB systems Li+ 22; (c, d) performance and impedance of LiFSI/6FDMH-DME series electrolytes 75;(e) structure-activity relationships of DME-FTEG molecular 76; (f) synthesis methods of DME-FTEG series moleculars 76.
Li+在电极界面需要进行去溶剂化过程才能到达电极界面发生反应,这个过程需要跨越势垒。不同电解液的溶剂化结构不同,因而去溶剂化能不同。Kim等78通过特殊电极、盐桥等手段,巧妙地平衡了诸如扩散、电极差异等影响电势的因素,使得所测得的电势能够反映不同电解液相对于标准电解液(DME)的溶剂化结构差异。LiFSI/FDMB电解液电势较低,代表去溶剂化能较高,有较多AGG溶剂化结构,这也是LiFSI/FDMB体系具有优异电池性能的重要原因。最近,Cui等79首次通过玻璃化冷冻手段,将含有电解液的SEI原位快速冻结在锂金属电极表面,通过冷冻电镜观测到更接近原位状态的SEI结构。研究人员发现,这种“湿态”的SEI与一般测试方法中“干态”的SEI具有明显的差异,“湿态”SEI具有明显更大的厚度,说明在原位状态下,SEI中含有大量的溶剂分子,从而导致SEI膨胀。但是,对于FDMB体系,“湿态”和“干态”SEI的厚度差别最小,说明该体系SEI的致密性和均匀性较好,能够更好地保护锂金属负极。
为了解决电解液动力学差的问题,该团队75于2021年将FDMB分子中的―CF2―链增长,构建出一系列分子如FDMH (图7d)。随着―CF2―链的增长,分子溶解锂盐的能力逐步下降,可以作为稀释剂与DME搭配而形成局部超浓电解液体系。LiFSI/DME/FDMH体系能够实现高库仑效率(99.5%)和良好的Li||NMC532电池性能(图8c),并且有极高的氧化稳定性(6 V vs. Li+/Li)。该体系在保持FDMB分子优良性能的同时,解决了FDMB阻抗较高的问题(图8d),这也说明稀释剂设计与部分氟代的分子设计方法有共通之处。
与上述方法类似,为了能在分子结构设计上同时满足溶剂分子和稀释剂的结构要求,该团队76构建了一系列分子,如DME-FTEG (图7e)等,其分子结构类似于两端为醚而中间为稀释剂的高度氟代醚结构。如图8e所示,DME-FTEG通过两端的―O―提供溶解盐的能力,而通过中间部分来起到抗氧化性和稳定结构的作用。而且改变端基―CH2O―的数量(图8f),可以对电解液性能进行更精确地调控和筛选。
3.2 端基碳链增长并进行F取代
众所周知,DME以其独特的分子结构可以类似于钳子与Li+配位,具有良好的溶解锂盐的能力。Bao和Cui团队77扬长避短,保持中间部分乙二醇的结构而延长两端碳链,构建了DEE分子(图7b)。该研究中4 mol·L−1LiFSI/DEE电解液,相较于4 mol·L−1LiFSI/DME,表现出了更强的抗氧化稳定性,匹配NMC正极的全电池在180圈循环后容量保持率高达~80%。从总体来看,增加溶剂碳链长度可以增加电解液的氧化稳定性:碳链增长导致溶剂结构变形性增大,空间位阻增大,不利于其与Li+配位,因而使得更多的阴离子出现在溶剂化鞘层。中间碳链的增长使得DME与Li+无法再形成五元环的配位结构,直接改变了溶剂与Li+的配位方式,但端基的增长却无此后果。而向其中引入F原子取代,其作用与稀释剂类似,但精确调控F原子的数量与位置可以优中选优。
Ma等80构建了一系列端基氟代醚E4F1、E4F2等(图7f、h),对端基上的氟取代进行了研究。其中间结构为不同数量的乙二醇脱水形成醚,端基为β-C连有―CF3或―CF2CF3。该设计中同样利用了醚基官能团能够使锂盐解离,同时氟化基团可以增强电解液氧化稳定性的策略。研究发现,该氟代醚分子具有较高的离子电导率(1.3 mS·cm−1),但离子电导率并不与醚链长度呈线性相关。另外,值得一提的是,研究发现这些电解质的氧化稳定性随着氟含量的减少而增加,这一现象在其他氟代醚中未被发现。
基于以上的理解,该团队23利用DEE的端基即为β-C的特点,对端基进行F取代以达到库仑效率、氧化稳定性和离子传导之间的平衡。该研究中作者根据溶剂分子上F原子数的不同命名为FXDEE(X = 3,4,5,6,图7g、i),X = 6时即为F完全取代端基C上的H原子。此类分子既是DEE的氟代衍生物,又在结构上与FDMB相似,两者看似不可协调的优点可以在同一个分子上实现,并达到了很好的平衡,同时实现了高电导率和低过电势。如图9a所示,在―CF3基础上构建了不对称的―CHF2,实现了更好的性能。正因如此,图9b中F6DEE并不是性能最好的溶剂,其循环过电位明显高于其他衍生溶剂;图9c中F4DEE和F5DEE氧化稳定性甚至高于F6DEE。在XPS分析中发现F3DEE体系、F6DEE体系中SEI表面存在SOxF,表明阴离子未完全分解或钝化。通过理论计算发现,―CHF2与―CF3相比与Li+作用更强,这可能是由于减少一个F原子使得参与配位的F原子更倾向于提供孤对电子,或者存在更小的空间位阻效应。同时,较多的F取代已经很好地保证了溶剂的抗氧化稳定性,因此如图9d中F4DEE体系与F5DEE体系的性能最为优异,F4DEE体系有稍好的传导动力学,而F5DEE体系有较好的循环性能。综上所述,通过对溶剂的精细调控,能够在高氧化稳定性、良好的溶解性与电导率之间达到很好的平衡(图9e)。
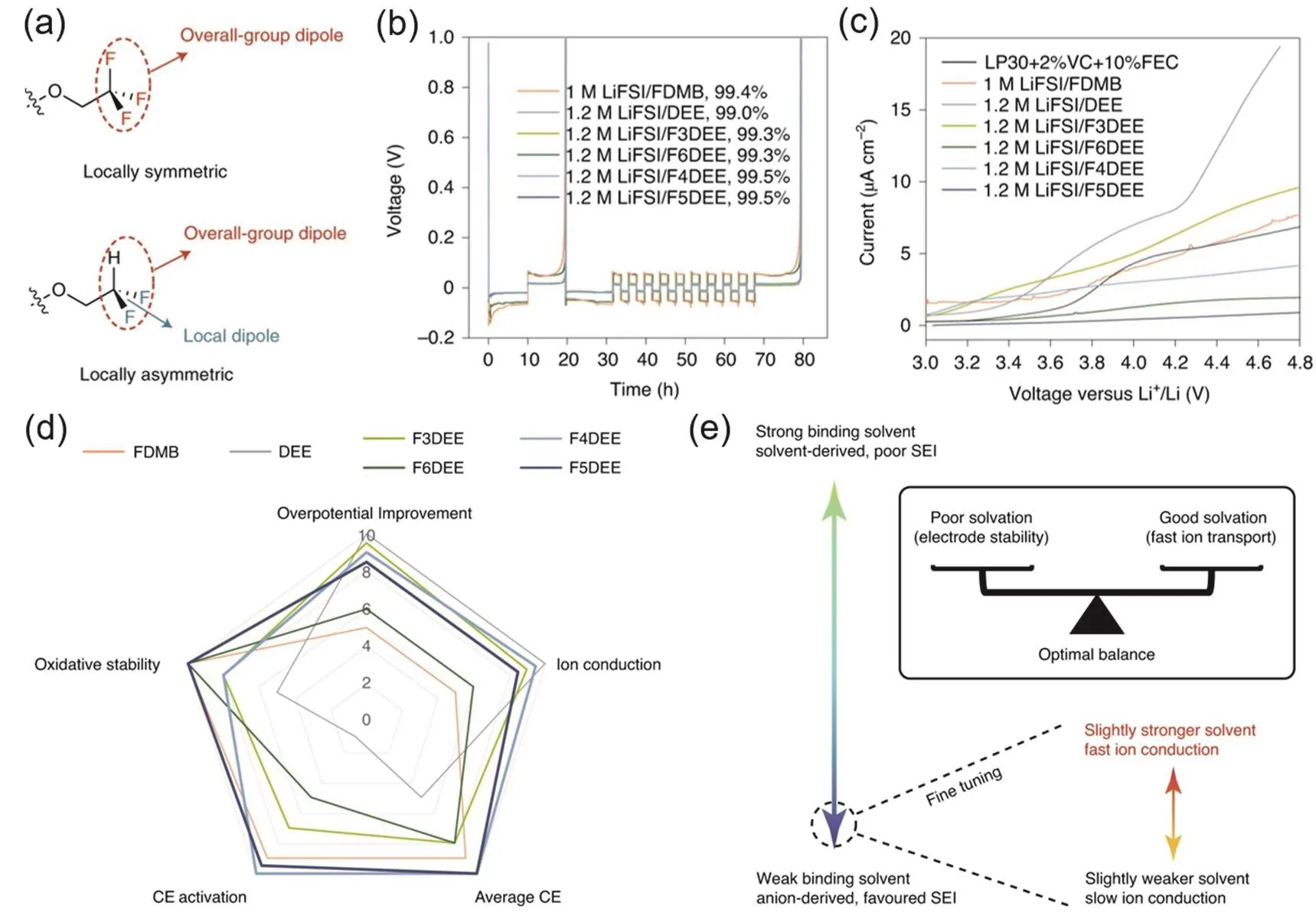
图9 (a) FXDEE系列分子中的―CF3基团和―CHF2基团;(b,c) FXDEE系列电解液的Li|Cu半电池性能与氧化稳定性;(d) FXDEE系列电解液性能对比;(e)电解液中氧化稳定性与溶解盐能力的平衡关系示意图23Fig. 9 (a) ―CF3 group and ―CHF2 group in FXDEE series molecules; (b, c) Li|Cu half-cell performance and oxidation stability of FXDEE series electrolytes; (d, e) schematic diagram of the equilibrium relationship between oxidative stability and ability to dissolve salts in electrolytes 23.
4 其他氟代分子
4.1 氟代磺胺分子
LiFSI81、LiTFSI82等锂盐能在电解液中大放异彩,离不开其阴离子的官能团氟代磺胺所具有的优点,即该官能团与Li+相互作用强度适中,具有良好的化学稳定性,分解产物稳定等。利用这种官能团进行分子设计,获得氟代磺胺分子作为溶剂,已在Li-S电池83和Li-O2电池84中得到了应用,并展现了良好的效果。Xue等85将该分子引入锂金属电池电解液中,利用LiFSI和FSA (N,N-二甲基氨基磺酰氟,图10a)组合成全氟代磺胺电解液。对于锂金属负极,其首圈库仑效率为91.1%,在10圈内迅速达到99%,而且能够很好地适配NMC622和LMO正极。2021年该团队37进一步研制出1 mol·L−1LiFSI/DMTMSA (1,1,1-三氟-N,N-二甲基甲磺酰胺,图10a)。如图10b所示,该电解液可以有效抑制在碳酸酯电解液体系中出现的锂枝晶、产气、正极过渡金属离子溶出、正极裂纹等问题。在该电解液中,电池放电容量超过230 mAh·g−1,100圈循环库仑效率超过99.65%;对于NMC 811正极甚至可以充电至4.7 V (vs. Li+/Li),90圈循环后容量保持率 大 于88%。 XPS数 据 分 析 表 明 , 1 mol·L−1LiFSI/DMTMSA电解液体系中形成的CEI含有更多的LiF无机成分和更少的有机成分。利用FXI的X射线吸收近边缘结构(XANES)模式,测量满电状态下正极中的Ni氧化态可以看出,1 mol·L−1LiFSI/DMTMSA体系中Ni的氧化态更高。该团队86还将1 mol·L−1LiFSI/DMTMSA匹配高电压(> 4.5 V,vs. Li+/Li)的LCO正极,当充电电压达到4.55 V时循环200圈后电池容量保持率高达89%,当充电到4.6 V时循环100圈后电池容量保持率为~85%。
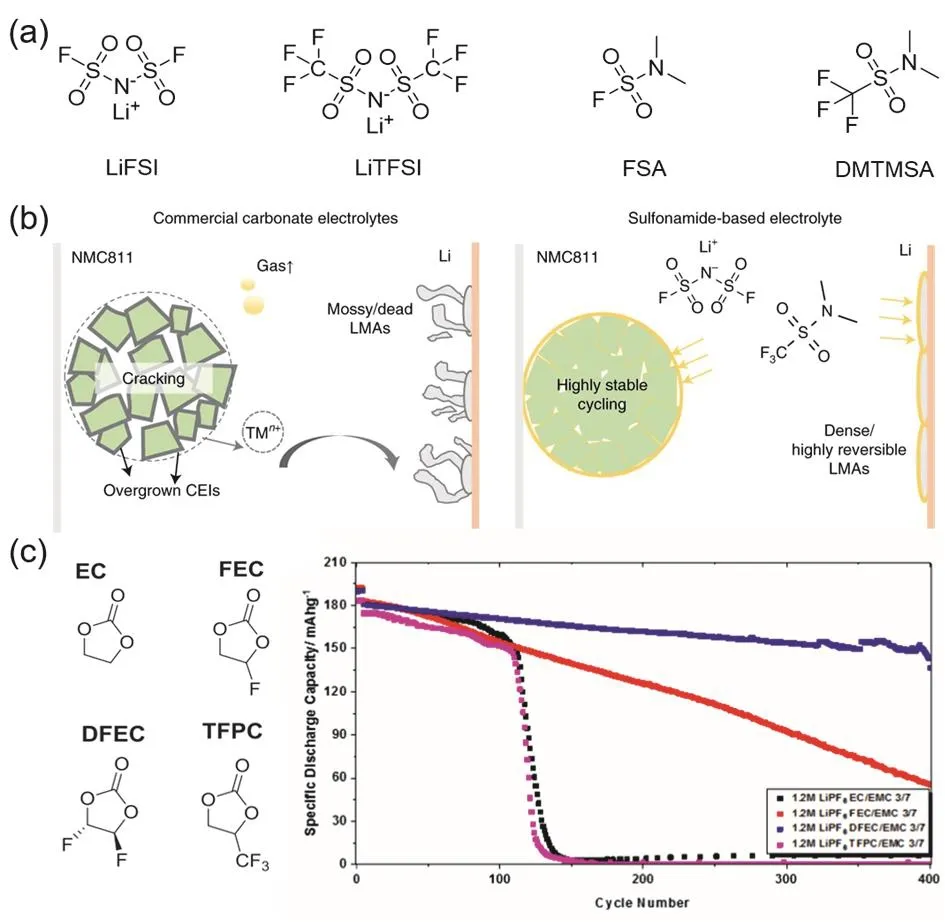
图10 (a)氟代酰胺盐与氟代酰胺溶剂分子结构;(b) DMTMSA电解液相比于碳酸酯电解液优势示意图37;(c)氟代环状碳酸酯分子结构与全电池性能88Fig. 10 (a) Molecular structure of fluorinated amide salt and fluorinated amide solvent; (b) schematic diagram of the advantages of DMTMSA electrolyte compared to carbonate electrolyte 37; (c) molecular structures of fluorinated cyclic carbonates and full cell test performance 88.
4.2 氟代碳酸酯分子
和醚类分子比较,碳酸酯分子在锂金属界面反应性更强,并且生成的SEI疏松多孔不稳定,库仑效率较低;但碳酸酯分子的抗氧化稳定性较强,可以匹配高电压正极材料,因此仍是十分重要的研究方向87,89,90。Zhang等87通过理论计算模拟,研究了不同位点F取代的直链碳酸酯溶剂对锂金属电池性能的影响。该研究的模拟结果表明,碳链长度对LiF产生具有重要影响,锂金属晶面对溶剂的反应活性也有重要作用。另外,锂盐的存在以及―CF3基团的数量与分布都是影响反应活性的重要因素。从总体来看,含有较少―CF3基团和较短氟化烷基链的直链碳酸酯溶剂可能有助于形成更多的LiF,生成富含LiF的SEI。
而对于环状碳酸酯EC,不同位置和数量的F取代也有不同效果。Su等88于2019年首次报道了DFEC (双氟代碳酸乙烯酯),是一种新型的稳定溶剂。与传统的碳酸乙烯酯相比,DFEC还原分解形成的稳定SEI显著了提升了锂金属电池的循环性能。如图10c所示,Li||NMC622电池体系在1.2 mol·L−1LiPF6/DFEC/EMC电解液中能够稳定循环400圈以上;但若将―CF3基团取代至EC上形成TFPC (4-三氟代甲基碳酸乙烯酯),其配制的电解液性能与EC组十分相近,无明显提高。而Zhang等91利用密度泛函理论(DFT)对FEC和DFEC两种溶剂分解路径进行模拟,发现两者的分解路径不同导致产生SEI组分也有差异。FEC的一种分解路径导致产生碳酸锂和氟代乙烯,另一种分解路径则导致产生一氧化碳和氟化锂。对于DFEC体系,理论模拟计算预测,一种机制导致碳酸亚乙烯酯和氟化锂的形成,另一种机制导致一氧化碳、氟化锂和乙二醛的形成。并且FEC分解倾向形成低聚合物,而DFEC倾向分解产生长链聚合物,因而FEC生成的SEI更加致密。Zhu等92也对EC、FEC、DFEC、TFEC等溶剂进行对比研究,用来揭示SEI的设计原理。研究发现由DFEC体系形成的SEI中LiF的含量最高(10.6%,原子分数),但TFEC体系形成的SEI中仅含有少量的LiF (1.7%)。并且由于分子结构中F原子数量的增加,DFEC分子和TFEC分子的反应性下降,溶剂分子分解不完全,导致所形成的SEI结构不够致密,因而在含有FEC的电解液中电池的综合性能最好。
最近文献也报道了将链状碳酸酯分子与环状碳酸酯联合使用,能够协同提升电池性能,例如上文中提到的1.28 mol·L−1LiFSI/FEC/FEMC/D2和0.7 mol·L−1LiBETI/FEC/DEC/M354。Su等93报道了三元电解液1.2 mol·L−1LiPF6/FEC/DFEC/FEMC (溶剂体积比3 : 3 : 14),该电解液匹配NMC811正极在3.0–4.4 V电压下能够稳定循环200圈,容量保持率> 90%。在该电解液体系中DFEC分解形成良好的SEI,而FEMC拥有优异的负极稳定性,并且通过FEC的溶剂化保护作用减轻FEMC分解产生的有害影响。Xiao等94开发了一种不易燃的高电压电解液1 mol·L−1LiPF6/FEC/BTFEC,该电解液设计中主要利用氟代碳酸酯为溶剂,大幅提升了电池性能。该电解液在Li|Cu半电池循环中表现了较高的库仑效率,300圈平均库仑效率为~98.8%。并且该电解液能够在−30 – 70 °C低温情况下正常运行,Li||NCM全电池在高电压4.7 V下能够稳定循环160圈,容量保持率为95.1%。
5 总结与展望
Li与F,分别位于元素周期表第一周期的两端,注定发生奇妙的反应35。将F引入到溶剂中的优点主要有:(1)提升电解液的氧化稳定性、阻燃性、浸润性等,使电池运行更安全;(2)电解液在正负极界面分解产生LiF,形成强健、均匀、致密的SEI和CEI,有效保护锂金属负极和高压正极界面95;(3)改变电解液中Li+的配位结构,产生独特的溶剂化鞘层,进而影响电池性能。综上所述,氟代溶剂对锂金属电池的作用明显,应用广泛。
F原子在溶剂分子中的位置与数量不同,会发挥出不同的作用:(1)将溶剂分子高度F取代,不能溶解锂盐,利用其稳定性和惰性形成稀释剂;(2)对于醚类溶剂而言,由于诱导效应,对α-C位置进行F取代会大幅降低Li+的溶解度,但对β-C位置进行F取代既可以保持一定的溶解度又能很好地发挥F的作用;但这对碳酸酯溶剂并不适用,α-C位置进行F取代对碳酸酯效果更佳;(3)溶剂分子在有一定数量F原子的前提下,对于端基C位置的F取代,―CHF2比―CF3表现出更好的动力学优势,且保持了溶剂分子良好的氧化稳定性。
锂金属电池的发展已经进入了崭新的阶段,不断有新的研究方法出现96、对机理研究愈发深入97、研究目标开始向CE > 99.9%逼近98,面向实用化的软包锂金属电池也进展迅速99。实现锂金属电池更高效、更安全的大规模商业化应用,是几代科研人的追求。而对溶剂进行氟取代,对溶剂结构进行合理设计无疑是一种行之有效的方法。虽然目前已开发出的氟代电解液种类并不多,但对电解液进行氟取代的作用与规律正在逐渐明晰,且溶剂的选择也多种多样,期待将来会有新型高效氟代溶剂的出现,使得锂金属电池真正走向大众。
猜你喜欢
特产研究(2024年1期)2024-03-12 05:40:56
云南化工(2021年11期)2022-01-12 06:06:18
核化学与放射化学(2020年4期)2020-08-21 07:45:08
山东冶金(2019年5期)2019-11-16 09:09:12
湿法冶金(2018年2期)2018-04-25 05:08:01
质谱学报(2016年4期)2016-08-02 05:33:25
电源技术(2016年2期)2016-02-27 09:04:59
中国资源综合利用(2016年7期)2016-02-03 03:00:19
分析测试学报(2015年9期)2015-12-17 16:44:27
应用化工(2014年7期)2014-08-09 09:20:28
