
CRISPR-Cas系统在细菌基因组编辑和代谢调控中的研究进展
2022-11-19 11:10肖雅丽张建华钟耀广
食品与发酵工业 2022年21期
肖雅丽,张建华,钟耀广*
1(上海海洋大学 食品学院,上海,201306)2(上海交通大学 农业与生物学院,上海,200240)
CRISPR-Cas系统,即规律成簇的间隔短回文重复序列(CRISPR基因座)及其相关蛋白(Cas基因簇),广泛分布于约50%的细菌和约90%的古菌中,是菌体内进化出的一种具有免疫记忆的获得性免疫防御系统,用于抵御外源遗传元件的入侵[1]。JINEK等[2]最早证明,来自化脓链球菌的II型CRISPR-Cas系统在体外可特异性切割双链DNA,将其用于基因组编辑,具有效率高、成本低、操作简单等优点。近年来,CRISPR技术的基本结构和作用机制逐渐被阐明,成为了继锌指核酸酶和类转录激活因子效应物核酸酶技术之后最新的基因组编辑技术,不仅可用于人类的基因治疗,还可以用于植物和动物的精确育种以及微生物工程,具有广阔的发展空间和应用前景。
如今,越来越多的CRISPR-Cas系统被发现和改造并用于细菌基因组编辑及基因表达调控,包括II型CRISPR-Cas9系统、V型CRISPR-Cpf1系统、内源I-B型和I-E型CRISPR-Cas系统。所有这些系统都有自己的特征蛋白、特异性的原始间隔区相邻基序(protospacer adjacent motif,PAM)等,可根据实验需求综合考虑,自由选择[3]。本文主要从CRISPR-Cas系统的结构、分类、作用机理,及其在细菌基因组编辑和基因表达调控等方面的应用进行综述,旨在为相关研究提供参考。
1 CRISPR-Cas系统的结构和免疫应答
1.1 CRISPR-Cas系统的结构
CRISPR基因座依次包括前导序列(Leader)、重复序列(Repeat)和间隔序列(Spacer)(图1)。
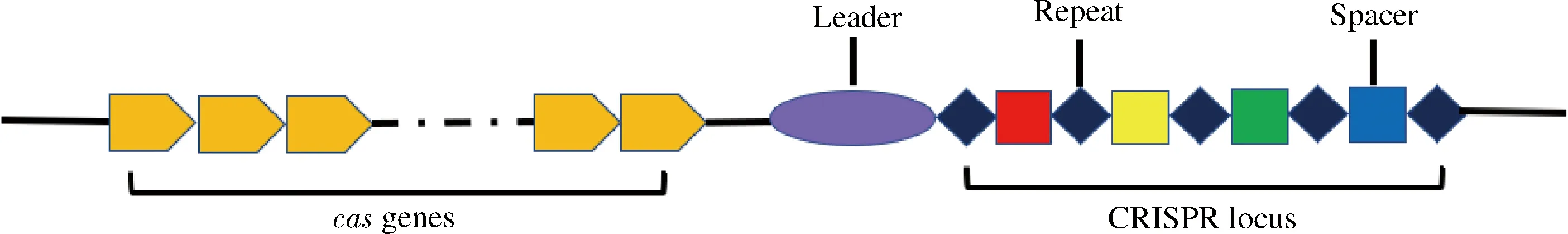
图1 CRISPR-Cas系统结构示意图[9]Fig.1 CRISPR-Cas system structure[9]
长度可达550 bp的前导序列多位于CRISPR基因座的5′端,直接与第一个重复序列相邻,富含A、T碱基。前导序列是添加新间隔序列的识别序列,相同的重复序列和特异的间隔序列交替排列于前导序列之后。同一个CRISPR基因座的重复序列高度保守,而不同CRISPR基因座的重复序列在序列和结构上均不相同[4]。入侵病毒或质粒的一个小片段(即原型间隔序列)被整合到CRISPR基因座中即为间隔序列,用于储存外来遗传元件的核苷酸信息。CRISPR基因座的两侧是大量极其多样化的cas基因,其编码的某些Cas蛋白具有核酸内切酶结构域、RNA和DNA结合域及参与转录调控的结构域,在CRISPR-Cas系统的适应性免疫中起着重要作用[5-7]。此外,Cas1和Cas2蛋白存在于大多数已知的CRISPR-Cas系统中,是将原型间隔序列插入CRISPR基因座所必需的[8]。
1.2 CRISPR-Cas系统的免疫应答
CRISPR-Cas系统的免疫应答过程如图2所示,包括3个主要阶段:适应(adaption)、表达(expression)和干扰(interference)[10]。从入侵的DNA/RNA中识别和提取原型间隔序列是CRISPR-Cas系统介导免疫的第一步,也称为适应阶段。首先,多功能Cas1-Cas2复合体识别入侵DNA上的PAM位点,并切割下入侵DNA的一部分(即原型间隔序列)。随后,原型间隔序列被整合到CRISPR基因座的前导序列和重复序列之间,成为间隔序列。也有一些CRISPR-Cas系统采用另一种适应机制——利用在CRISPR基因座上编码的逆转录酶进行逆转录,从而从入侵的RNA中获取间隔序列[11-12]。

图2 CRISPR-Cas系统免疫应答3阶段[16]Fig.2 Immune process of CRISPR-Cas system[16]
在表达阶段,CRISPR基因座中的间隔重复序列通常被转录为较长的CRISPR RNA前体(pre-crRNA)。在多亚基效应复合物中的核酸内切酶或者核糖核酸酶Ⅲ(RNase Ⅲ)和反式激活CRISPR RNA(tracrRNA)的介导下,pre-crRNA被加工成短的成熟CRISPR RNA(crRNA),每个成熟crRNA分子都包含一段间隔序列和一部分重复序列。成熟的crRNA分子与效应蛋白或多亚基效应复合物结合,形成crRNA-效应复合物[13]。
最后,当宿主细胞受到相同的外来DNA/RNA入侵时,系统发生干扰,成熟crRNA引导Cas蛋白特异性识别PAM位点后,靶向切割入侵DNA或RNA[14-15]。
2 CRISPR-Cas系统的分类和作用机制
2.1 CRISPR-Cas系统的分类
随着对CRISPR-Cas系统研究的深入,各类不同的CRISPR-Cas系统逐渐被发现,根据系统效应子的不同将其分为2大类:第1类CRISPR-Cas系统的效应子是由多个Cas蛋白组成的多亚基效应复合物;第2类系统的效应子为单一的多域Cas效应蛋白。MAKAROVA等[10]于2020年根据Cas效应蛋白的不同和多亚基效应复合物的差异将CRISPR-Cas系统进一步分为6种类型:第1类包括Ⅰ型、Ⅲ型和Ⅳ型;第2类包括Ⅱ型、Ⅴ型和Ⅵ型。
第1类系统中,Ⅰ型系统的多亚基效应复合物通常包含1个Cas6亚基、1个Cas5亚基、几个Cas7亚基和特征蛋白。Ⅰ型系统还编码解旋酶Cas3,它通常与HD(His-Asp)核酸酶结构域融合。Ⅲ型系统组成类似于Ⅰ型,不同的是,Ⅲ型系统的特征蛋白为Cas10,与HD(His-Asp)核酸酶融合的也是Cas10。与Ⅰ型和Ⅲ型系统差别较大的Ⅳ型CRISPR-Cas系统通常位于质粒上,缺乏cas1、cas2基因以及目标DNA切割酶的编码基因cas3或cas10,但拥有编码Cas5 (Csf3)、Cas7 (Csf2)和特征蛋白(Csf1)的基因。根据特征蛋白的不同,Ⅰ型系统进一步细分为I-A到I-F和I-U 7个亚型。I-A到I-D型的特征蛋白分别为:Cas8a、Cas8b、Cas8c、Cas10 d;I-E型的特征蛋白为Cse1和Cse2;I-F型的特征蛋白为Csy1、Csy2、Csy3和Csy6;I-U型的特征蛋白未知[3,17]。
骆马湖局开展采砂管理工作以来,河湖采砂从曾经的混乱无序到如今的逐步规范有序,采砂管理从高压严打到寻求源头管理的标本兼治,从流域与区域当初的“一事一议”到建立长效的合作机制,这一切,都归功于不断推进的流域性河湖采砂联合管理机制。但是,这种联合管理机制目前还不是十分完善,仍然存在着以下几个方面急需解决的问题。
在第2类系统中,Ⅱ型、Ⅴ型、Ⅵ型的根本区别在于其效应蛋白的结构不同。Ⅱ型系统的效应蛋白是Cas9,包含2个高度保守的核酸酶结构域:HNH(His-Asn-His)核酸酶结构域和RuvC核酸酶结构域[18]。Ⅴ型系统包括V-A到V-I和V-U 10种亚型,只有V-A型被用于细菌基因组编辑,效应蛋白是Cas12a(Cpf1),由1 200~1 300个氨基酸组成,仅包含一个RuvC核酸酶结构域[3,19]。VI型系统的效应蛋白为Cas13(也称C2c2),包含2个高等真核生物和原核生物核苷酸结合结构域,与任何已知的DNA核酸酶结构域均不同源,靶向RNA[20]。
2.2 CRISPR-Cas系统的作用机制
上述I-VI型的CRISPR-Cas系统中,IV型CRISPR-Cas系统通常位于质粒上,缺乏从入侵DNA/RNA上提取原型间隔序列所需的Cas1和Cas2以及靶向切割入侵DNA/RNA所需的核酸内切酶[17];Ⅵ型CRISPR-Cas系统只能靶向RNA[20]。下面重点介绍可以靶向切割DNA的Ⅱ型CRISPR-Cas9、Ⅴ型CRISPR-Cpf1和I型CRISPR-Cas系统的作用机制。
2.2.1 Ⅱ型CRISPR-Cas9系统的作用机制
Ⅱ型CRISPR-Cas9系统包含Cas9蛋白、crRNA和tracrRNA。tracrRNA是由重复序列转录而来,负责激活RNase Ⅲ以促进pre-crRNA的加工,生成成熟的crRNA。成熟的crRNA与tracrRNA和Cas9形成复合物,并引导Cas9识别目标DNA中的PAM位点(5′-NGG-3′)。随后,HNH核酸酶结构域切割外源DNA中与间隔序列互补的单链,RuvC核酸酶结构域切割另一条单链,使DNA双链断裂(double stranded break,DSB),产生平末端。将crRNA与tracrRNA分子进行适当改造,整合为一条RNA链,即单链引导RNA(single guide RNA,sgRNA),可以同时发挥crRNA和tracrRNA的功能[9]。
Cas9核酸酶产生的DSB可以通过非同源末端连接途径或同源定向修复系统进行修复。多数细菌缺少非同源末端连接途径,需依赖DNA模板介导的同源定向修复途径完成修复,否则DSB很容易引起细胞死亡[21]。YAO等[22]人为构建了一个CRISPR-Cas9系统(带有sgRNA和目标基因的同源修复臂)并转入宿主菌中,该系统识别宿主菌基因组上的目标基因并进行切割,最后通过同源修复臂与基因组上的目标位点发生同源重组而实现基因的缺失、突变或插入。
2.2.2 V型CRISPR-Cpf1系统的作用机制
Cpf1是继Cas9之后的一种新型核酸酶,与Cas9只识别5′-NGG-3′这一PAM位点不同,它能识别富含T碱基的PAM位点(5′-YTN-3′和5′-TTTN-3′)[3],扩展了可靶向识别的基因组DNA范围。此外,CRISPR-Cpf1是目前最简单的CRISPR-Cas系统,只包含crRNA和Cpf1蛋白。Cpf1不需要tracrRNA的参与就可以将pre-crRNA加工成成熟的crRNA,成熟的crRNA与Cpf1形成复合物并靶向识别目标DNA。然后,Cpf1蛋白依次切割非互补链和互补链以产生DSB。最后,通过同源定向修复或者非同源末端连接途径修复DNA断裂片段。因此,通过设计由间隔序列(23~25 nt)和同向重复序列(19 nt)组成的单链向导crRNA来引导Cpf1识别和切割目标DNA并提供同源修复臂修复DSB即可实现细菌的基因组编辑。而且,CRISPR-Cpf1产生黏性末端,与CRISPR-Cas9(产生平末端)相比具有更高的同源重组修复效率,可更简单高效地编辑细菌基因组[23]。
2.2.3 I型CRISPR-Cas系统的作用机制
3 CRISPR-Cas系统在细菌基因组编辑中的应用
依赖于锌指核酸酶和类转录激活因子效应物核酸酶的基因组编辑技术存在耗时且效率低、需要进行选择性标记、难以插入大DNA片段以及大片段的删除和插入会对下游基因造成影响等问题[25]。CRISPR-Cas基因组编辑技术具有快速精确、无标记、简便高效等优势,因此被广泛应用于微生物研究。目前,用于细菌基因组编辑的主要有II型CRISPR-Cas9系统、Ⅴ型CRISPR-Cpf1系统、内源Ⅰ型CRISPR-Cas系统以及由CRISPR-Cas9衍变而来的CRISPR-Cas9n系统。
3.1 CRISPR-Cas9介导的基因组编辑
CRISPR-Cas9是最早被用于细菌基因组编辑的CRISPR-Cas系统。随着CRISPR技术的发展,有学者[26-27]于2016年开发了一种基于CRISPR-Cas9系统的快速简便的基因组编辑方法,只需要构建一个质粒便可以在3 d内实现大肠杆菌的基因组编辑,而用传统的基因组编辑技术大约需要7 d。CRISPR-Cas9不仅缩短基因组编辑周期,还可提高编辑效率。RONDA等[28]利用CRISPR-Cas9系统和λ-Red重组酶在大肠杆菌中同时编辑3个基因,重组效率达到了96.5%~99.7%。相比之下,传统重组系统的效率仅为0.68%~5.40%。
此外,CRISPR-Cas9系统还可编辑更大的DNA片段。SU等[29]将CRISPR-Cas9系统和λ-Red重组系统结合,成功地将12 kb的DNA片段整合到大肠杆菌W3110的染色体中,编辑效率可达100%。SYNEFIARIDOU等[30]在肺炎链球菌D39V中利用CRISPR-Cas9系统实现了长达24 kb的染色体大片段删除。
3.2 CRISPR-Cas9n介导的基因组编辑
Cas9蛋白造成的双链断裂有时会导致宿主细胞死亡。为解决这一问题,研究人员通过突变Cas9蛋白的HNH结构域(H840A)或RuvC结构域(D10A)得到了Cas9切口酶:Cas9n (Cas9 Nickase)。Cas9n只 能切割DNA的一条链,切口容易修复且脱靶率低。当修复系统无法修复由Cas9引入的DSB时,可尝试用Cas9n代替Cas9[31]。例如,GOH等[32]使用CRISPR-Cas9n系统在嗜酸乳杆菌NCFM、加氏乳杆菌ATCC 33323和副干酪乳杆菌Lpc-37中进行基因组编辑,实现了300 bp~1.9 kb的染色体缺失,编辑效率为35%~100%。ZHOU等[33]开发了基于CRISPR/Cas9n的基因编辑工具,不仅在恶臭假单胞菌(Pseudomonasputida)中敲除了不同长度(98~4 643 bp)的DNA片段,而且敲入了长度从1.9 kb到15 kb不等的DNA片段。同样,CRISPR-Cas9n介导的基因组编辑也可有效缩短操作周期。用同源重组双交换法敲除干酪乳杆菌的单基因至少需要24 d。为了能快速、精确地编辑干酪乳杆菌基因组,SONG等[34]建立了CRISPR-Cas9n(D10A)系统,将编辑时长缩短至9 d,单基因敲除效率最高可达65%。
3.3 CRISPR-Cpf1介导的基因组编辑
继CRISPR-Cas9之后,具有结构简单、应用范围广、基因组编辑效率高等优点的CRISPR-Cpf1系统被开发和应用。LI等[35]使用CRISPR-Cpf1和同源定向修复系统成功敲除了天蓝色链霉菌的actI-orf1和redX基因,单基因编辑效率为90%~95%,证明了该系统的简单高效,可用于多种基因组工程。ZHAO等[36]将CRISPR-Cpf1系统和核酸外切酶-重组酶(RecET)系统结合起来,实现了谷氨酸棒杆菌ATCC 14067基因组的大片段缺失。其中,1 kb和20 kb基因缺失的编辑效率分别为79.6%和36.4%。WU等[37]改造新弗朗西斯菌(Francisella)U112的CRISPR-Cpf1系统并将其用于枯草芽孢杆菌,以100%的效率实现了单基因插入、双基因敲除和多点突变(最多6个)。同年,HAO等[38]借助CRISPR-Cpf1系统以高编辑效率精确地实现了枯草芽孢杆菌的基因敲除,其中包含一个38 kb的超大基因簇。
3.4 内源Ⅰ型CRISPR-Cas介导的基因组编辑
第2类CRISPR-Cas系统中的CRISPR-Cas9/Cpf1是强大的基因组编辑工具,然而Cas9或Cpf1作为一种异源蛋白很难被引入某些宿主菌中,从而难以进行基因组编辑。据报道,大多数的CRISPR-Cas系统属于第一类,且Ⅰ型最为常见[5]。因此,可以借助宿主菌体内的I型系统来实现基因组工程,即构建内源I型CRISPR-Cas系统,利用内源的Cas蛋白靶向切割宿主菌基因组上的目标基因。
PYNE等[39]设计了内源I-B型CRISPR-Cas系统,成功删除了巴氏梭菌中的cpaAIR基因,编辑效率高达100%。而相比之下,用Ⅱ型CRISPR-Cas9系统编辑该基因的效率仅为25%。ZHANG等[40]使用丁酸梭菌的内源I-B型CRISPR-Cas系统成功地进行了pyrF和spo0A双基因敲除,编辑效率为100%。ZHOU等[41]报道了在酪酸梭菌中利用内源I-B型CRISPR-Cas系统进行基因组编辑,成功敲除了spo0A和aldh基因,效率高达100%。这些结果表明内源CRISPR-Cas系统是细菌基因组编辑的有效工具,有巨大的发展潜力。
4 CRISPR-Cas系统在细菌基因转录调控中的应用
4.1 CRISPR-dCas9系统介导的基因转录调控
虽然已经开发出了对宿主细胞低毒性的CRISPR-Cas9n系统,但在一些DNA修复效率较低且外部修复机制无效的菌株中,Cas9n仍可能导致细胞死亡[42]。所以,科研人员制备了Cas9蛋白HNH核酸酶结构域(H840A)和RuvC核酸酶结构域(D10A)的双突变体,即灭活核酸内切酶:dCas9。CRISPR-dCas9系统不会引发DNA双链或单链的断裂,可在不引起DNA损伤的前提下识别目标DNA并干扰基因的转录[43]。dCas9蛋白既可以与转录激活因子融合以产生CRISPR激活(CRISPRa)系统[44],也可以与特定基因组位点结合从而抑制下游基因的转录,在sgRNA的引导下选择性地调节目标基因的表达,称为CRISPR抑制(CRISPRi)系统[45]。
传统的RNA干扰技术可用于下调目标基因表达,但仅限于具有适当宿主机制的特定生物体,并表现出显著的脱靶效应和细胞毒性[46]。在细菌中,CRISPRi是比RNA干扰更好的基因转录调控工具,能有效、可逆地抑制多种细菌中目标基因的表达,如谷氨酸棒杆菌中的pyc、gltA、idsA和glgC[47]、铜绿假单胞菌(Pseudomonasaeruginosa)的prtR[48]以及金黄色葡萄球菌的tarO、tarH和tarG[49]。将CRISPR-dCas9系统应用于工业微生物可有效地提高目标产物的产量,如琥珀酸[50]、乙醛酸[51]、白藜芦醇[52]、1,4-丁二醇[53]等。相比之下,关于细菌CRISPRa的报道则较少。
4.2 CRISPR-ddCpf1介导的基因转录调控
由CRISPR-Cas9改造而来的CRISPR-dCas9系统虽然已广泛应用于基因的转录调控,但对于多个靶点需要多个sgRNA的独立表达。相比之下,采用CRISPR-Cpf1系统则只需设计由19 nt的同向重复序列和23~25 nt的间隔序列组成的crRNA即可实现单基因敲除。更重要的是,将多个crRNA直接串联,只需要一个启动子驱动即可简单高效地实现多基因敲除。而且,crRNA序列的长度比CRISPR-Cas9系统的sgRNA短约60 nt,大大降低了构建系统的难度和成本[3,35]。同时,Cpf1蛋白同时具有DNA内切酶(DNase)和RNA内切酶活性,且DNase失活并不影响RNA内切酶的活性,系统依然能生成成熟的crRNA,靶向识别目标DNA[54]。于是,ZHANG等[55]将Cpf1中的第993位谷氨酸突变为丙氨酸,获得了DNase失活的DNase-dead Cpf1(ddCpf1),并将其成功用于大肠杆菌的多位点转录调控。LI等[35]开发了基于ddCpf1的CRISPRi系统用于多重基因抑制,在天蓝色链霉菌中同时抑制了3种抗生素生物合成基因(cpkA、redX、orf1)的表达,效率约为70%。
4.3 内源I-E型CRISPR-Cas系统介导的基因转录调控
通过敲除负责降解目标DNA的cas3,大肠杆菌中的内源性I-E型CRISPR-Cas系统被用作可编程基因表达调节器[56]。CHANG等[57]在大肠杆菌中构建了内源I-E型CRISPR-Cas系统,将大肠杆菌中的靶基因表达量下调了82%。TARASAVA等[58]使用改良的内源I-E型CRISPR-Cas系统,可以同时靶向6个不同的基因,通过筛选增加丙二酰辅酶A通量的突变体,提高了大肠杆菌中3-羟基丙酸的产量。
5 总结及展望
自JINEK等[2]于2012年在体外证明了CRISPR-Cas9系统的DNA切割机制后,该系统凭借高效、经济、简便等优点逐渐代替了传统的基因组编辑技术,被广泛应用于细菌的基因组编辑。结构更简单的CRISPR-Cpf1系统拓宽了CRISPR-Cas系统的应用范围并且具有更高地基因组编辑效率。而针对无法引入Cas9和Cpf1的宿主菌,可利用细菌的内源I型CRISPR-Cas系统进行基因组编辑和代谢调控。同时,由于Cas9造成的DSB容易引起细胞死亡,于是在其基础上开发出了CRISPR-Cas9n系统。另外,对Cas9和Cpf1进行改造得到的dCas9和ddCpf1还可以用于调控基因的转录表达。
但CRISPR-Cas系统依然存在改进的空间。首先,CRISPR-Cas系统对PAM位点的特异性识别以及宿主菌对异源蛋白的排斥限制了该系统的应用范围。因此,未来应努力改造Cas蛋白以扩大其识别范围并继续开发内源CRISPR-Cas系统。其次,细菌基因组编辑效率因物种而异,表明宿主菌的生理活动会影响CRISPR-Cas系统。今后应开发更可靠、可诱导和广泛适用的表达结构,使Cas效应子和gRNA在不同宿主菌中均能稳定表达。最后,随着CRISPR-Cas系统的大量应用,将生成包含数百万甚至数十亿变体的大型文库[59],因此开发高通量技术很有必要,例如高效转化方法、机器人平台和微流体系统等。相信随着科学技术的进步,CRISPR-Cas系统将来会拥有更优异的性能。
猜你喜欢
农业工程学报(2022年8期)2022-08-08
湖北农业科学(2022年11期)2022-07-18
作物学报(2022年9期)2022-07-18
小学生学习指导(低年级)(2019年3期)2019-04-22
中文信息(2017年2期)2017-04-13
江苏农业科学(2016年11期)2017-03-21
智能计算机与应用(2016年4期)2016-09-26
安徽理工大学学报·社会科学版(2014年6期)2015-02-06
读写算·小学低年级(2014年4期)2014-07-24
银行家(2014年1期)2014-02-14
