
高效液相色谱一测多评法评价乳核散结片10种成分质量*
2022-11-15 08:06薛艳丽娄朝晅薛小琦田广林
医药导报 2022年11期
薛艳丽,娄朝晅,薛小琦,田广林
(1.河南科技大学第一附属医院药学部,洛阳 471000;2.河南科技大学法医学实验教学中心,洛阳 471000;3.遂成药业股份有限公司,新郑 451100)
乳核散结片收载于《中华人民共和国药典》2020年版一部,由当归、淫羊藿、郁金、柴胡、漏芦等10味中药组方而成,具有疏肝活血、祛痰软坚功效,临床主要用于肝郁气滞、痰瘀互结所致数量不同、大小不一、质软或中等硬的乳房肿块或结节,或乳房胀痛、经前疼痛加剧的乳腺增生病[1-3]。
乳核散结片现行质量标准[4]仅对佐药淫羊藿中一种成分进行定量研究,未对方中君药和臣药所含成分进行研究分析,笔者也未检索到与本品质量分析相关的文献报道。笔者参考中药质量标志物研究方法,从整体上把握中药指标性成分,采用高效液相色谱一测多评[5-6](high performance liquid chromatography-quantitative analysis of multi-components by single-marker,HPLC-QAMS)法对乳核散结片方中君药郁金所含活性成分双去甲氧基姜黄素、去甲氧基姜黄素和姜黄素,臣药当归所含活性成分洋川芎内酯I、洋川芎内酯H、阿魏酸松柏酯、藁本内酯和欧当归内酯A,佐药淫羊藿所含主要活性成分淫羊藿苷和宝藿苷I含量进行同时测定,以期为全面研究和评价乳核散结片质量提供参考。
1 仪器与试药
1.1仪器 高效液相色谱仪(安捷伦公司,Agilent 1200型;岛津公司,LC-20AT型);液相色谱柱(Agilent TC-C18柱、Diamonsil C18柱和Hypersil gold C18柱,规格均为4.6 mm×250 mm,5 μm);XS105DU型电子分析天平(梅特勒-托利多公司,感量:0.01 mg);KQ-250E型超声波清洗器(昆山市超声仪器有限公司)。
1.2试药 乳核散结片(广州白云山中一药业有限公司,规格:每片0.36 g,批号:S00015、S00023、S00024);色谱级乙腈,其余试剂均为分析纯;淫羊藿苷、宝藿苷I、双去甲氧基姜黄素、去甲氧基姜黄素和姜黄素对照品(中国食品药品检定研究院,批号分别为110737-201516,111852-201603,112004-201501,112003-201501,110766-201721,含量分别为94.2%、99.9%、95.0%、98.5%、98.7%);洋川芎内酯I、洋川芎内酯H、阿魏酸松柏酯和藁本内酯A对照品(上海同田生物技术股份有限公司,批号分别为15102222,17092822,17112023,17111023,含量分别为98.5%,99.6%,94.4%和98.1%);欧当归内酯A对照品(成都普瑞法科技开发有限公司,批号:PRF10021443,含量:97.7%)。
2 方法与结果
2.1对照品溶液的制备 精密称取各对照品适量,70%甲醇溶解,制成各对照品浓度分别为0.198,0.112,5.764,4.156,0.638,4.952,2.734,0.798,1.072和1.986 mg·mL-1混合对照品贮备液;精密吸取混合对照品贮备液1.0 mL,70%甲醇定容至20 mL,制成各对照品浓度分别为9.9,5.6,288.2,207.8,31.9,247.6,136.7,39.9,53.6和99.3 μg·mL-1的对照品溶液。
2.2供试品溶液的制备 取除去薄膜衣的乳核散结片20片,研细,取细粉2 g,精密称定,置25 mL量瓶,加70%甲醇适量,超声处理30 min,放冷经70%甲醇定容,摇匀得乳核散结片供试品溶液。按乳核散结片质量标准中工艺处方制备无当归、无淫羊藿和无郁金阴性供试品,再按上述方法制成各阴性供试品溶液。
2.3色谱条件及专属性实验 色谱柱:Agilent TC-C18柱(4.6 mm×250 mm,5 μm),柱温30 ℃;流动相乙腈-0.6%醋酸溶液,梯度洗脱(0~11.0 min,19.0%乙腈;11.0~24.0 min,19.0%→38.0%乙腈;24.0~32.0 min,38.0%→50.0%乙腈;32.0~48.0 min,50.0%→60.0%乙腈;48.0~60.0 min,60.0%→19.0%乙腈),流速1.0 mL·min-1;检测波长为280 nm(0~32.0 min检测洋川芎内酯I、洋川芎内酯H、阿魏酸松柏酯、藁本内酯、欧当归内酯A、淫羊藿苷和宝藿苷I)[7-14]和420 nm(32.0~60.0 min检测双去甲氧基姜黄素、去甲氧基姜黄素和姜黄素)[15-18];进样量10 μL。依法进样“2.1—2.2”项各溶液检测(图1),洋川芎内酯I等10种成分峰形对称,理论板数按各成分色谱峰计均≥5500,且与相邻色谱峰的分离度均>1.5,阴性供试品对乳核散结片中10种成分测定未产生干扰。
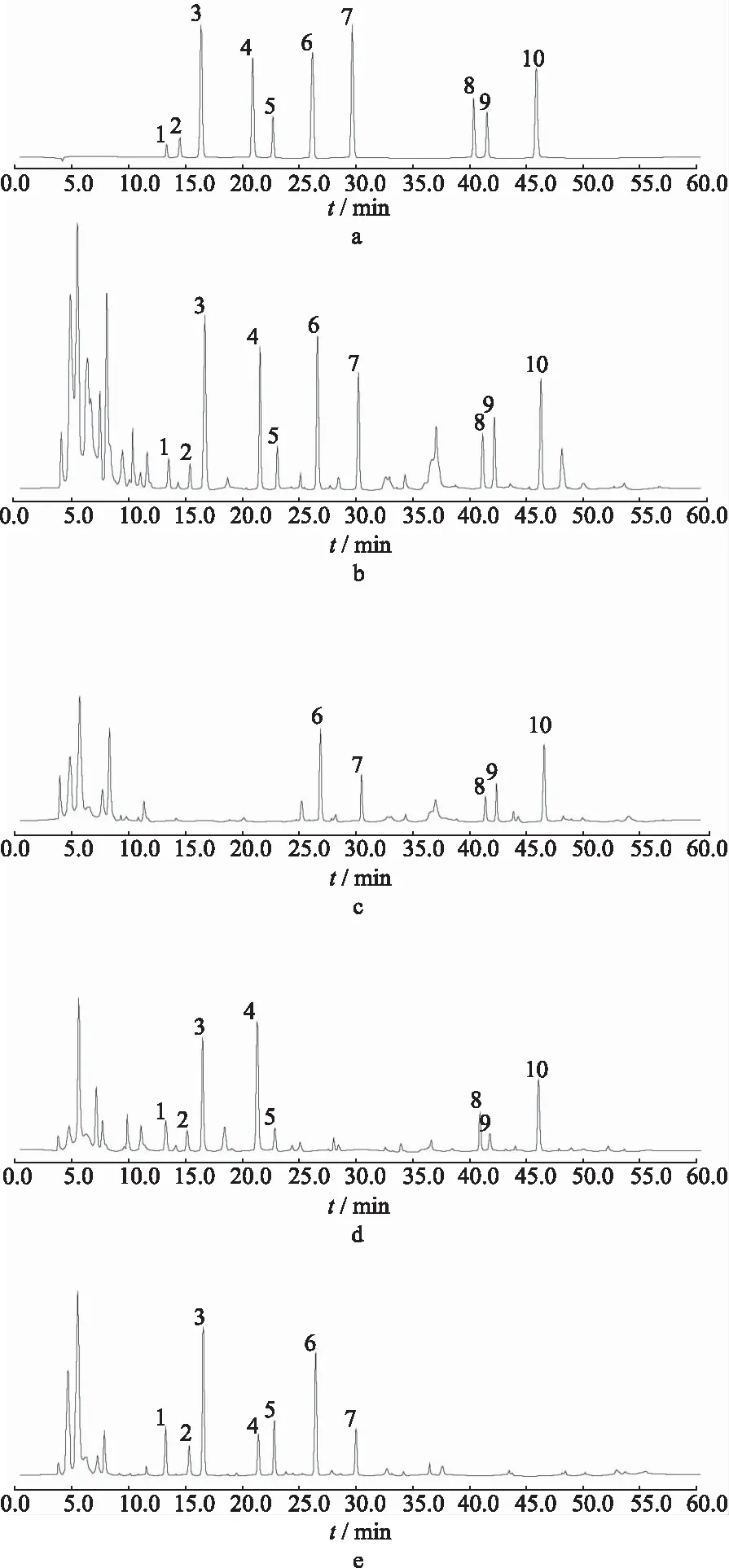
1.洋川芎内酯I;2.洋川芎内酯H;3.阿魏酸松柏酯;4.藁本内酯;5.欧当归内酯A;6.淫羊藿苷;7.宝藿苷I;8.双去甲氧基姜黄素;9.去甲氧基姜黄素;10.姜黄素;a.混合对照品溶液;b.乳核散结片样品溶液;c.当归阴性样品溶液;d.淫羊藿阴性样品溶液;e.郁金阴性样品溶液。
2.4线性关系考察 分别精密吸取“2.1”项混合对照品贮备液0.1,0.2,0.5,1.0,2.0和4.0 mL,分别以70%甲醇定容至20 mL,得6个混合对照品溶液,依法测定,以洋川芎内酯I等10种成分浓度对峰面积绘制标准曲线,得10种成分回归方程(表1)。

表1 乳核散结片中各成分标准曲线、线性范围与相关系数
2.5精密度实验 连续进样“2.1”项下对照品溶液6次,得洋川芎内酯I等10种成分峰面积RSD分别为1.03%,1.19%,0.52%,0.67%,0.98%,0.59%,0.74%,0.86%,0.72%和0.61%。
2.6重复性实验 将同一批乳核散结片按“2.2”项方法制备供试品溶液6份,依法进样得洋川芎内酯I等10种成分含量RSD分别为0.81%,1.34%,1.40%,1.72%,0.96%,1.58%,1.34%,1.40%,1.72%和1.63%。
2.7乳核散结片供试品溶液稳定性考察 取乳核散结片供试品溶液1份,于制备后0,2,4,6,12和24 h进样检测,得洋川芎内酯I等10种成分峰面积的RSD分别为1.33%,0.91%,0.99%,1.25%,0.57%,1.08%,0.91%,0.99%,1.25%,0.95%,表示乳核散结片供试品溶液24 h内稳定。
2.8加样回收率实验 取已知洋川芎内酯I等10种成分含量的乳核散结片(批号:S00015),除去薄膜衣后研细,取9份,每份1 g,精密称定,置25 mL量瓶,分别精密加入加标对照品溶液(洋川芎内酯I、洋川芎内酯H、阿魏酸松柏酯、藁本内酯、欧当归内酯A、淫羊藿苷、宝藿苷I、双去甲氧基姜黄素、去甲氧基姜黄素、姜黄素对照品浓度分别为0.154,0.078,4.582,3.192,0.462,3.854,2.138,0.612,0.794和1.512 mg·mL-1)0.8,1.0和1.2 mL,各平行3份,再按“2.2”项方法制备加样供试品溶液。依法分析检测,得洋川芎内酯I等10种成分平均加样回收率及RSD分别为97.85%(0.82%),96.97%(1.03%),100.07%(0.62%),99.42%(1.34%),98.64%(0.80%),99.43%(1.13%),100.02%(0.67%),98.35%(0.97%),98.70%(1.23%)和97.99%(1.65%)。
2.9相对校正因子的测定及耐用性考察
2.9.1相对校正因子的测定 依法进样“2.4”项6个混合对照品溶液,检测洋川芎内酯I等10种成分的峰面积,以淫羊藿苷为内参物,按照相对校正因子计算公式:k/s=k/s=(Xk×Ys)/(Xs×Yk)(式中X为质量浓度,Y为峰面积,s为其他成分,k为内参物)计算其他9种成分相对校正因子(表2)。
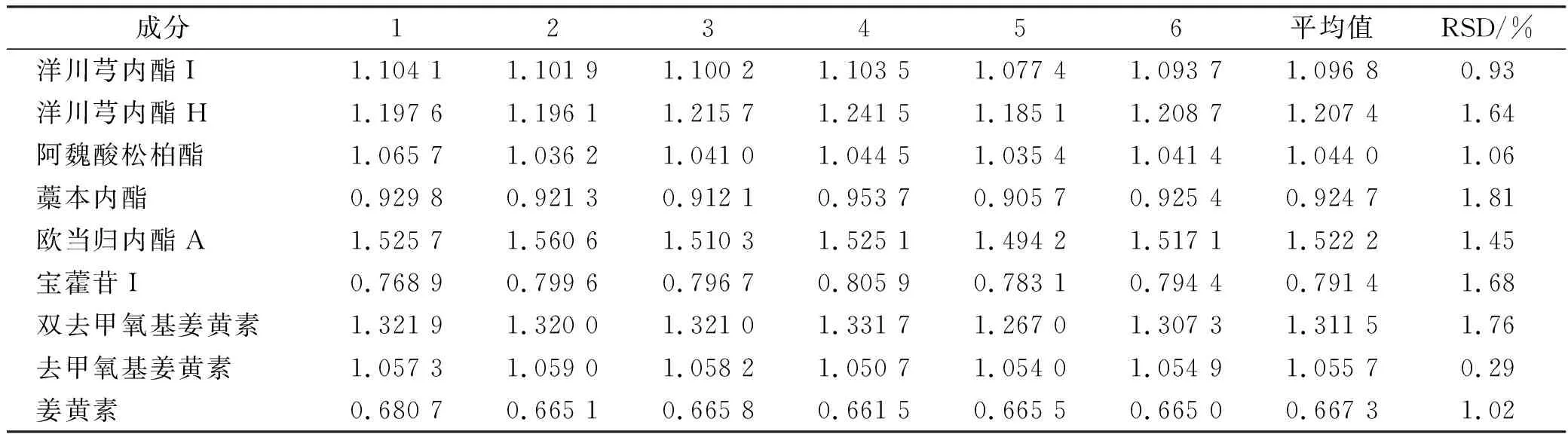
表2 各成分相对校正因子
2.9.2仪器和色谱柱对相对校正因子的影响 依法进样“2.1”项下对照品溶液,考察Agilent 1200型和LC-20AT型高效液相色谱仪及Agilent TC-C18柱、Diamonsil C18柱和Hypersil gold C18柱条件下测得的相对校正因子(表3)。
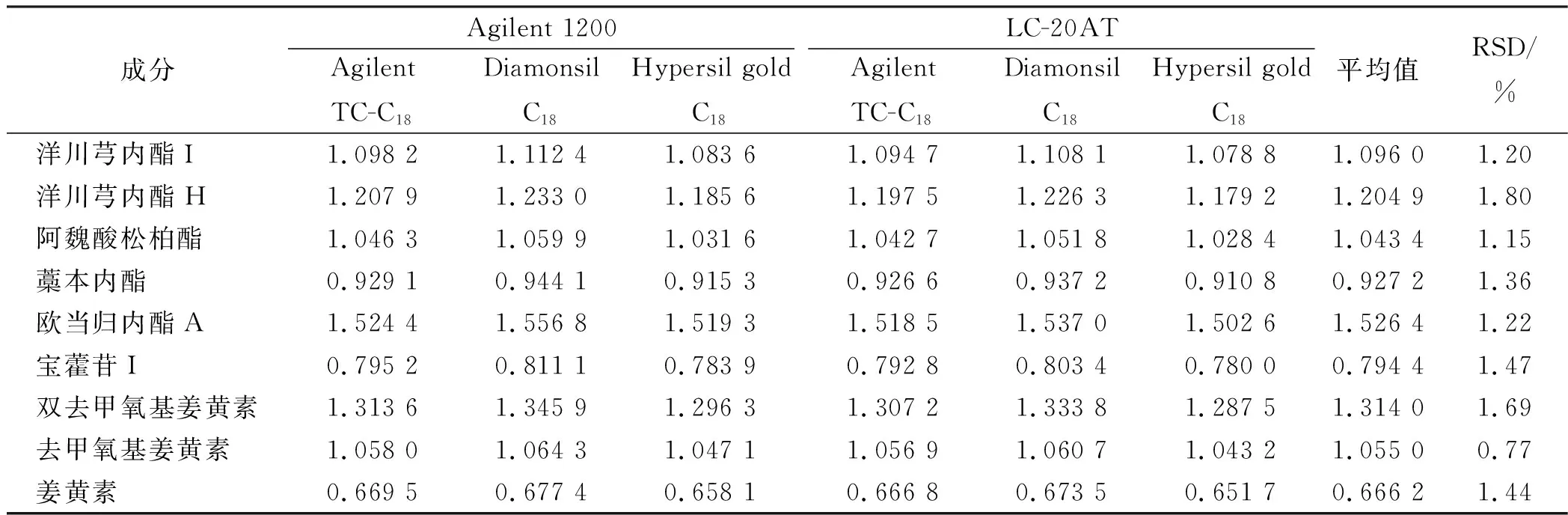
表3 不同仪器和色谱柱测得的相对校正因子
2.9.3柱温对相对校正因子的影响 依法进样“2.1”项下对照品溶液,考察不同柱温下测得的相对校正因子(表4)。
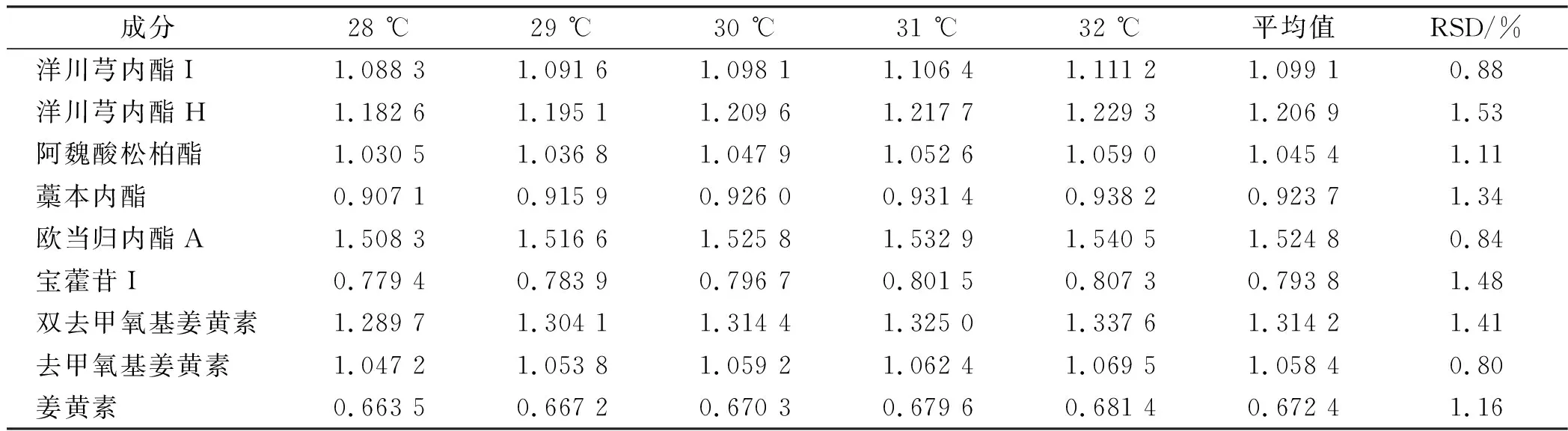
表4 不同柱温下测得的相对校正因子
2.9.4流速对相对校正因子的影响 依法进样“2.1”项下对照品溶液,考察不同流速条件下测得的相对校正因子(表5)。
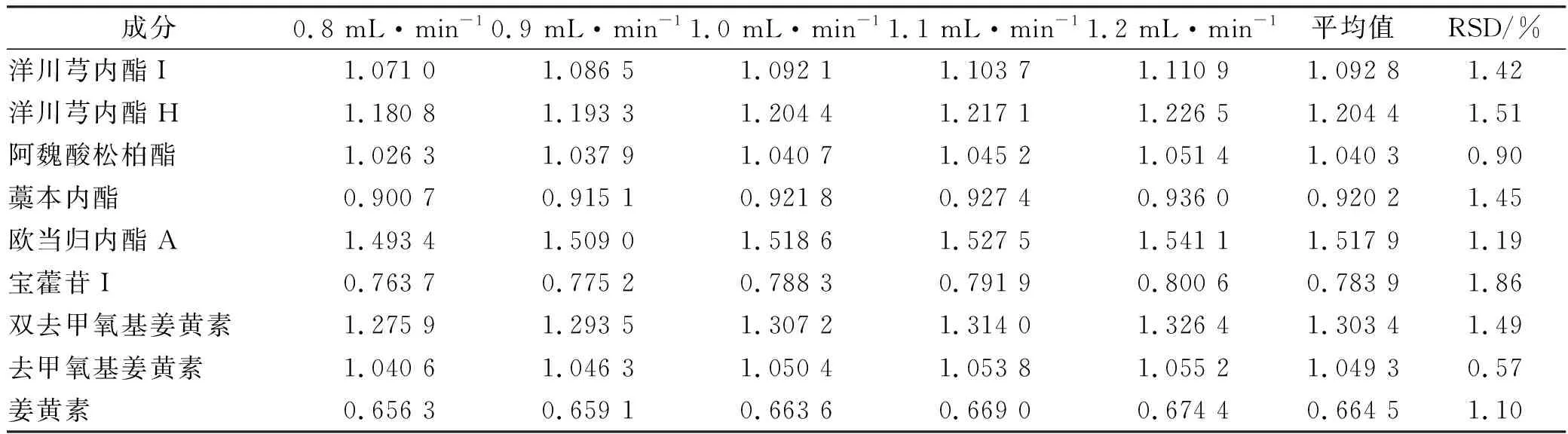
表5 不同流速下测得的相对校正因子
2.10色谱峰的定位 依法进样“2.1”项对照品溶液,检测,记录各成分保留时间,采用相对保留时间值法对洋川芎内酯I等10种成分进行色谱峰定位,考察高效液相色谱仪和液相色谱柱对相对保留时间值的影响(表6)。
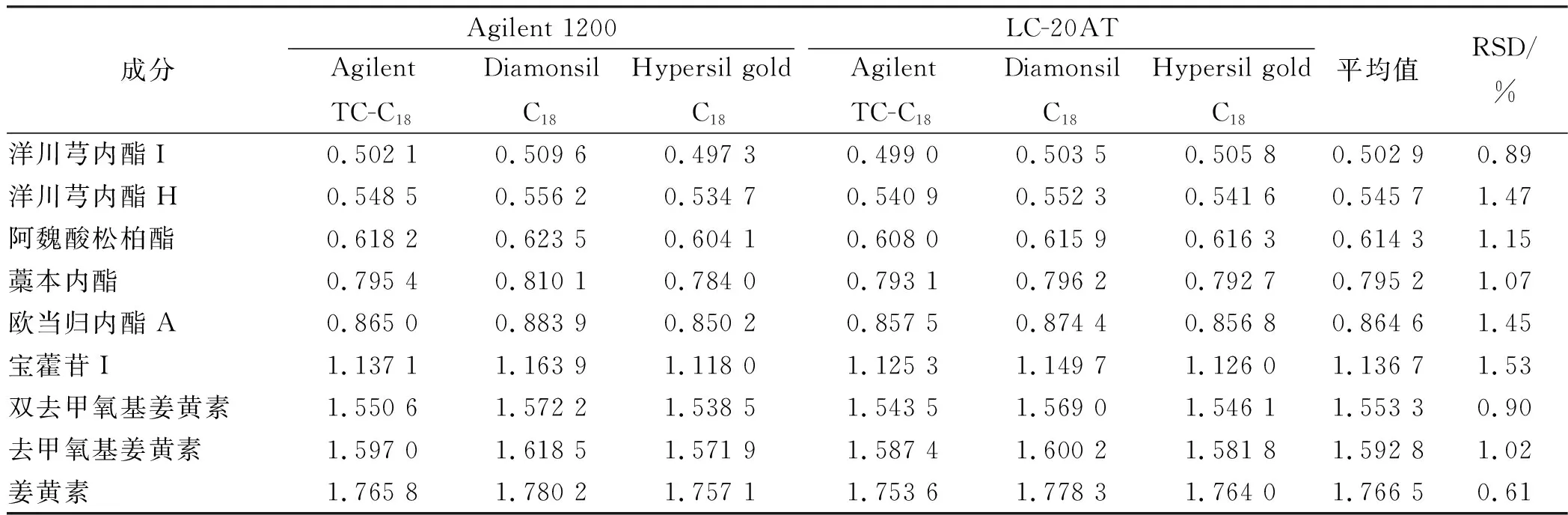
表6 不同仪器和色谱柱条件下待测成分色谱峰相对保留时间值
2.11两种方法测定结果比较 取乳核散结片3批,制备乳核散结片供试品溶液,依法进样测定,先用外标法计算10种成分含量,再用“2.9.1”项相对校正因子值计算其他9种成分含量(表7)。结果一测多评法计算值与外标法结果差异无统计学意义(RAD<2.0%)。
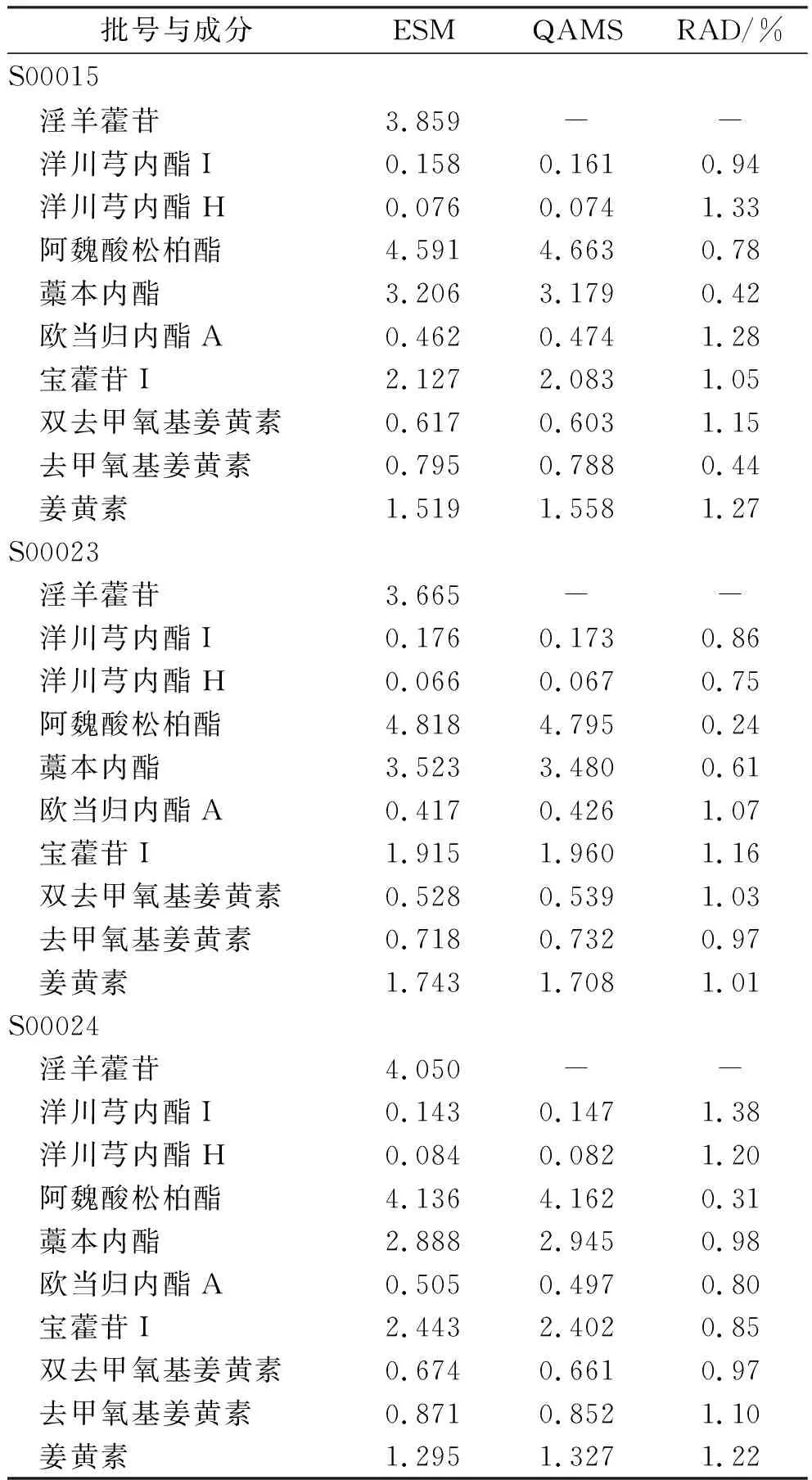
表7 乳核散结片各成分含量测定结果
3 讨论
3.1流动相的选择 乳核散结片为复方制剂,方中成分多且杂,本实验在选择流动相时,首先对比甲醇-水和乙腈-水两个基础流动相对基线平稳性及各成分分离效果的影响,结果发现甲醇-水流动相体系检测时,基线严重漂移,个别成分无法准确积分;乙腈-水流动相体系则基线水平,不足之处是阿魏酸松柏酯A、淫羊藿苷色谱峰出现拖尾,与相邻峰分离不完全,且峰对称度不符合要求。对乙腈-甲酸溶液[7-9,15]、乙腈-醋酸溶液[10,16-18]进行对比,又经过摸索酸溶液浓度,最终确定流动相为乙腈-0.6%醋酸溶液[18]进行梯度洗脱,乳核散结片中洋川芎内酯I、洋川芎内酯H、阿魏酸松柏酯、藁本内酯、欧当归内酯、淫羊藿苷、宝藿苷I、双去甲氧基姜黄素、去甲氧基姜黄素、姜黄素与相邻色谱峰均能有效分离,色谱峰峰形尖锐,对称性好。
3.2供试品溶液制备方法的确定 本实验在确定制备供试品溶液方法时,以乳核散结片中洋川芎内酯I等10种成分的提取效果为首选指标,同时考虑杂质干扰因素,以水[7]、70%甲醇[8]、甲醇[9]和稀乙醇[11-12]为溶剂,采用超声提取[7-13,16-17,19]和加热回流提取[18]两种提取方式,提取时间20,30,40和60 min时制得的溶液进样检测,最终优选确定供试品溶液制备方法为70%甲醇超声提取30 min。
HPLC因检测自动化、灵敏、应用范围广、检测快速、效率高、供试品制法简便等优点而被广泛应用于各种药物及其制剂的分析研究,笔者在本实验采用HPLC-QAMS法对乳核散结片中10种成分进行含量同步测定,实验进行了含量方法学验证及校正因子耐用性考察,对比两种方法含量测定结果,方法易操作,灵敏、准确、可靠,可以指导制药企业在生产过程中优化质控参数,为全面评价乳核散结片产品质量,确保产品质量均一安全提供实验依据。
猜你喜欢
现代食品(2022年11期)2022-12-06
九江学院学报(自然科学版)(2022年2期)2022-07-02
现代食品科技(2022年5期)2022-05-30
今日农业(2021年2期)2021-11-27
包装与食品机械(2021年5期)2021-11-06
健康之家(2021年19期)2021-05-23
家庭百事通·健康一点通(2021年1期)2021-02-24
今日农业(2020年23期)2020-12-31
Digital Chinese Medicine(2020年1期)2020-09-25
文萃报·周五版(2018年25期)2018-08-13
