
功能型离子液体协同吸收NH3和CO2的密度泛函理论研究
2022-11-13 07:32朱先会王甫夏杰成袁金良
化工学报 2022年10期
朱先会,王甫,夏杰成,袁金良
(宁波大学海运学院,浙江 宁波 315832)
引 言
CO2捕集与封存(CCS)是我国应对气候变化的重要战略选择,也是我国履约减排任务、应对未来挑战的重要技术选择之一[1]。以氨水为吸收剂的氨法碳捕集技术具备高CO2吸收容量和低再生能耗等优势,且不存在设备腐蚀、氧化降解等问题,成为近年来国内外“低能耗碳捕集”的一个重要研究方向。然而,由于NH3的高挥发特性,NH3逃逸问题仍是氨法碳捕集工艺中的一大技术壁垒[2]。因此,开发合适的添加剂来抑制NH3的挥发显得尤为重要。
离子液体(ILs)作为一种新型绿色溶剂,具有极低的挥发性、不会造成气相污染、低损耗、解吸能耗低、常温下可以溶解大多数气体、稳定性好、性质可调等优势,在气体脱除领域获得了广泛的关注[3-5]。利用ILs 的低挥发性、低饱和蒸气压以及高稳定性进行NH3的吸收可以解决传统水洗法脱除NH3存在的再生能耗高、废水量大以及氨精馏困难等问题[6]。陈晏杰等[7]提出了以[C4mim][BF4]为吸收剂的NH3吸收与多级闪蒸回收工艺,其流程模拟结果显示NH3的回收率高达93.3%。Zeng等[8]研究发现金属ILs与硅胶结合的多孔材料可以大量吸附NH3。Xu 等[9]通过实验发现ILs 添加剂可以降低氨法碳捕集中NH3的逃逸。Shi 等[10]通过分子模拟研究发现阳离子对NH3的吸收起主导作用,主要原因是[Emim]阳离子与NH3之间形成了较强的氢键。Shang 等[11]根据ILs的结构可调性设计合成了三种含有不同数量氢键的离子液体([Bmim][NTf2]、[Bim][NTf2]、[HOOC(CH2)3mim][NTf2]),实验研究结果显示,[Bim][NTf2]具有最高的NH3吸收容量(超过2.69 mol NH3·(mol ILs)-1)。曾少娟等[12]通过总结发现羟基功能化的ILs比普通的ILs可以吸收更多的NH3,主要在于羟基与NH3形成了较强的氢键。
进一步地,ILs 在吸收CO2方面也呈现出了较大的潜力。Blanchard 等的研究表明CO2在[Bmim][PF6]中具有很高的溶解度[13],随后对多种常规ILs溶解度的研究证实了其对CO2具有较高的选择性[14]。在大多数ILs 吸收CO2的过程中,阴离子发挥了重要作用[15],主要表现在CO2在ILs 中的物理溶解和ILs 与CO2弱相互作用的结合[16],以及ILs 在水中的溶解度和亲和力。同时,一些含有特定化学反应的特殊基团被设计引入到传统的ILs 中,以开发出具有低蒸气压、低再生能耗、抗氧化能力强和高吸收容量等特性的功能型ILs[17]。比如,一些研究学者借鉴有机胺吸收CO2的原理,将氨基引入到ILs 的阳离子上,从而极大地改善了ILs吸收CO2的能力[18-19]。
虽然ILs 在吸收CO2和NH3方面均表现出了良好的效果,但由于ILs 对CO2和NH3吸收作用方式的不同,目前研究的ILs 难以同时兼顾CO2和NH3的吸收效果[9]。同时,ILs 对CO2和NH3的吸收能力均受ILs中阴、阳离子的极性,酸碱性,密度,黏度,以及功能基团等因素的影响[20],需对ILs的结构和功能基团进行设计与优化,以达到对NH3和CO2的协同吸收效果。基于此,本文根据文献中ILs 与NH3和CO2作用时最可能的结构以及ILs 阴、阳离子对CO2和NH3作用方式的不同,设计五种功能型ILs,利用密度泛函理论对其结构进行优化及频率计算,探讨ILs 单独吸收NH3和CO2及协同吸收的作用机理以及吸收能力。
1 计算方法
密度泛函理论(density functional theory, DFT)是一种研究多电子体系电子结构的方法。DFT在物理和化学等领域都有广泛的应用,特别是在研究分子和凝聚态的性质等方面,是计算化学领域最常用的方法之一。本研究采用DFT 中的B3LYP∕6-31’++G(d,p)基组研究ILs 构型及其与气体分子之间的相互作用。电子密度拓扑分析采用Multiwfn 软件包[21]来实现,其他所有的计算均依据Gaussian 09[22]程序完成。
1.1 相互作用能的计算
相互作用能是离子对与其相应的阴、阳离子之间的能量差,由式(1)进行计算得到。在相互作用能的计算中,通常会有基组重叠误差(base set superposition error, BSSE)产生,导致计算出的相互作用能偏大。因而,在研究分子间的弱相互作用时,需要消除基组重叠误差的影响。利用Boys 等[23]提出的理论,由式(2)进行基组叠加误差计算。进一步地,通过式(1)和式(2)可推导出式(3)所示的ILs离子间相互作用能的计算方法。
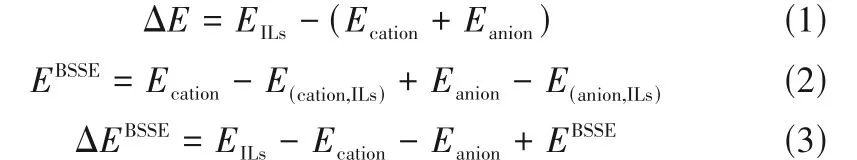
式中,EILs是阴阳离子对的电子能量;Ecation是阳离子的电子能量;Eanion是阴离子的电子能量;EBSSE是基组叠加误差。
在DFT 理论6-31 G(d,p)基组条件下,首先对五种ILs 的构型进行预优化,通过ILs 相互作用能的计算得到离子对较为稳定的构型。然后选用更复杂的6-31’++G(d, p)基组进行几何构型优化,以获得能量最低的稳定结构。
1.2 AIM理论分析
基于优化后的ILs 构型,通过Bader 分子中的原子理论(AIM)[24],在Multiwfn 软件中进行电子密度拓扑理论分析。AIM 方法已被证明可用于ILs 体系中的氢键研究[25-26]。通过计算可得到电子密度、拉普拉斯电子密度值,并用式(4)得到氢键结合能(EHB,hydrogen bonding energy),以此来分析形成的氢键强弱。同时,使用Multiwfn 可以分析静电势(ESP)、原子偶极矩以及原子电荷,以预测亲电和亲核最可能的攻击位置,为ILs 与气体分子的相互作用提供理论基础。

2 结果与分析
2.1 离子液体的构型优化及相互作用能
在B3LYP 杂化泛函6-31’++G(d,p)基组水平上对 设 计 的 五 种ILs([HEMim][Glu]、[HEMim][Asp]、[HEBim][Asp]、[HEBim][Ala]、[HEBim][His])进 行 结构优化。这五种ILs 由两种羟基化的咪唑型阳离子和四种氨基酸阴离子组合而形成不同的空间结构。图1 所显示的是能量最低、最稳定的结构。经过结构优化和频率分析证实各ILs 均无虚频,可以稳定存在。其中咪唑环上的二面角N15-C1-N16-C2 和N16-C2-C3-N15 分别为0.1195°和0.1123°,表明咪唑环上的各原子基本处在同一个平面上;咪唑环上的原子与其周围原子的二面角大都在179.6°左右,表明咪唑环与其周围各个原子也基本处在同一个平面内,这为ILs自身的稳定提供了重要支持。
根据价键理论,当两原子间距小于相应两原子之间的范德华半径之和而大于共价键键长,并且其相应角度大于90°时则认为形成了氢键。从图1 可以看出,这五种ILs 内部形成了较多的氢键,这为离子液体的稳定性提供了基本保证。根据文献中的结果:氢键应该有相对较大的电子密度(ρBCP)和拉普拉斯电子密度(∇2ρBCP)。其中ρBCP用来表示原子之间成键的强弱,ρBCP越大形成的氢键就越强。∇2ρBCP是ρBCP的二阶导数,可以用来判断原子之间的成键类型。当∇2ρBCP<0,则表明键点(BCP)处的电荷是积聚的,相邻的两个原子之间以共价键的形式存在;相反,如果∇2ρBCP>0,则表明BCP 处的电荷是发散的,相邻两个原子之间以氢键、离子键等闭壳层作用存在。一般来说,ρBCP的取值范围为0.002~0.04 a.u.,∇2ρBCP的取值范围为0.02~0.15 a.u.。然而这只是传统氢键的取值范围,有一些氢键的ρBCP和∇2ρBCP的取值范围会比传统氢键的取值范围大。表1列出了五种ILs 的ρBCP和∇2ρBCP以及氢键作用能(EHB),由此可判断五种ILs 内部氢键的形成及强弱。例如:[HEMim][Asp]中形成氢键的∇2ρBCP都为正值,H 原子和O 原子形成的化学键的键长为1.74~2.28 Å(1 Å=0.1 nm),ρBCP的范围为0.013~0.045 a.u.,符合形成氢键的判据且形成了三个氢键,其中C1—H6…O28 处的氢键最强,其氢键结合能为38.45 kJ·mol-1。
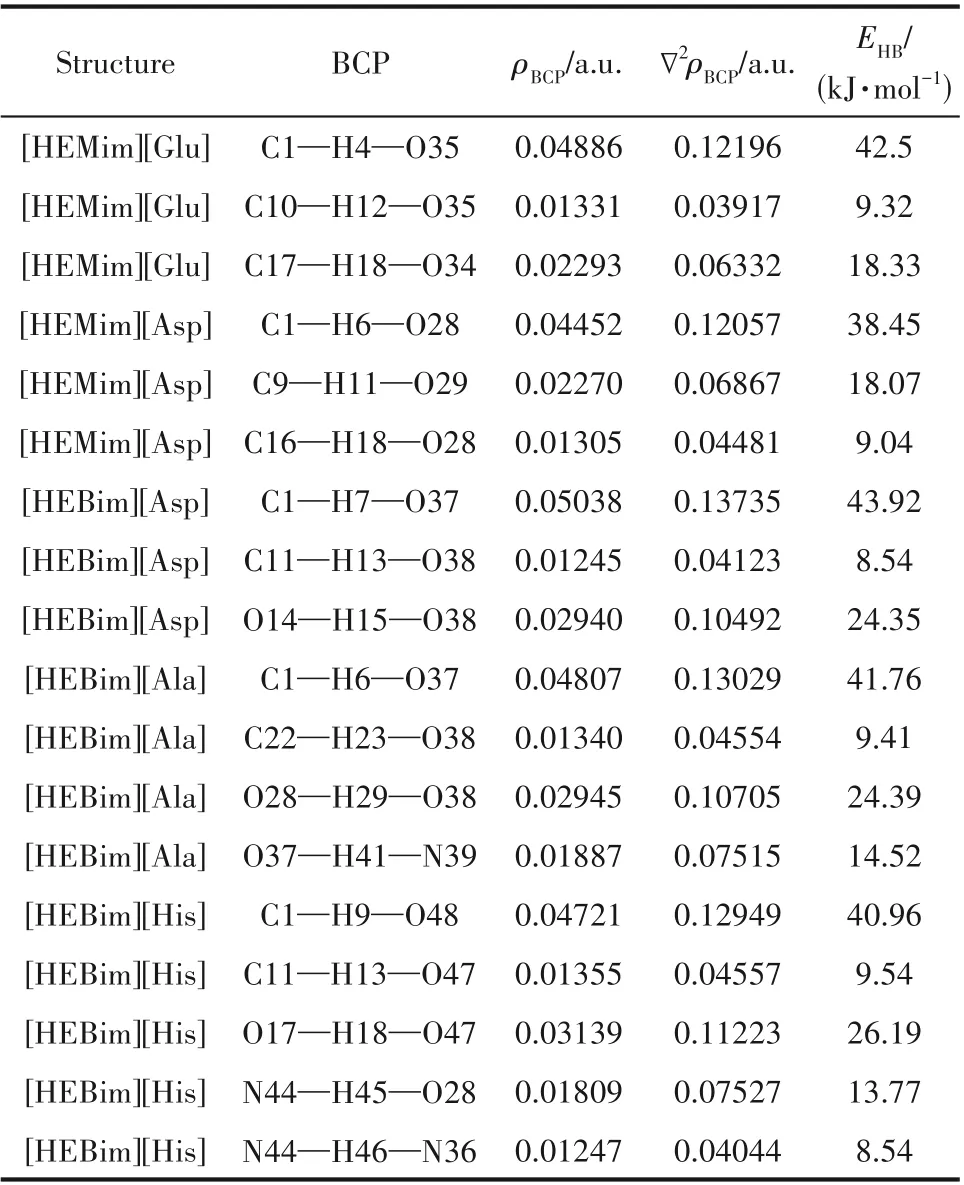
表1 在B3LYP/6-31’++G(d,p)基组水平下计算得到的五种离子液体的电子密度性质Table 1 Properties of electron density of BCP of five ionic liquids calculated at B3LYP/6-31’++G(d,p)level

图1 在B3LYP∕6-31’++G(d,p)基组水平下优化得到的五种离子液体较为稳定的构型Fig.1 Optimized structures of five ionic liquids at B3LYP∕6-31’++G(d,p)level
图2 列出了所设计的五种ILs 的零点作用能以及经过BSSE 校正的相互作用能。从图中可以看出,五种ILs 的零点作用能和经过BSSE 校正的相互作用能在-421~-390 与-416~-388 kJ·mol-1之间。与张营[27]研究的[C3mim] [Glu] 的相互作用能(-350.91~-306.42 kJ·mol-1)相比,本研究的ILs 相互作用能的绝对值更大。这是因为ILs 功能化之后,阴阳离子之间形成了更多的氢键,使得其稳定性更好。通过以上分析可以看出[HEBim][His]最稳定,其经过BSSE 校正之后的相互作用能达到-415.73 kJ·mol-1。
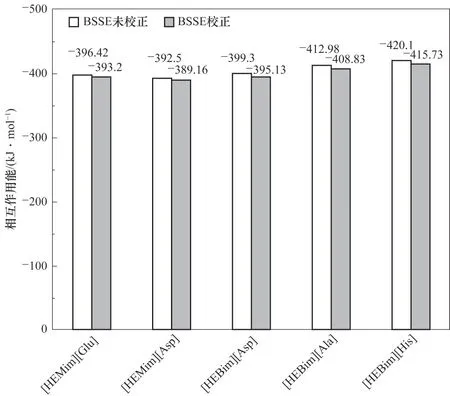
图2 在B3LYP∕6-31’++G(d,p)基组水平下计算得到的五种离子液体的相互作用能Fig.2 Interaction energies for five ionic liquids at B3LYP∕6-31’++G(d,p)level
2.2 离子液体的静电势分析与原子电荷分析
静电势分析可以更好地理解ILs 与气体分子之间的相互作用机理,找出ILs 与气体相互作用的最佳位点。图3 是五种ILs 的静电势能面示意图,ILs表面的蓝色和红色分别对应的是正静电势和负静电势(ESP),其中颜色越深代表静电势越强。从图中可以看出,正电势大多集中在咪唑环附近,其中咪唑环上的H 原子正电势最强,阳离子羟基中的H 原子正电势次之。而负静电势大多集中在阳离子的O原子以及阴离子的O原子和N原子附近。上述结果说明咪唑环上的H 原子和羟基中的H 原子较易与NH3相 互 作 用,O 原 子 和N 原 子 较 易 与CO2相 互作用。
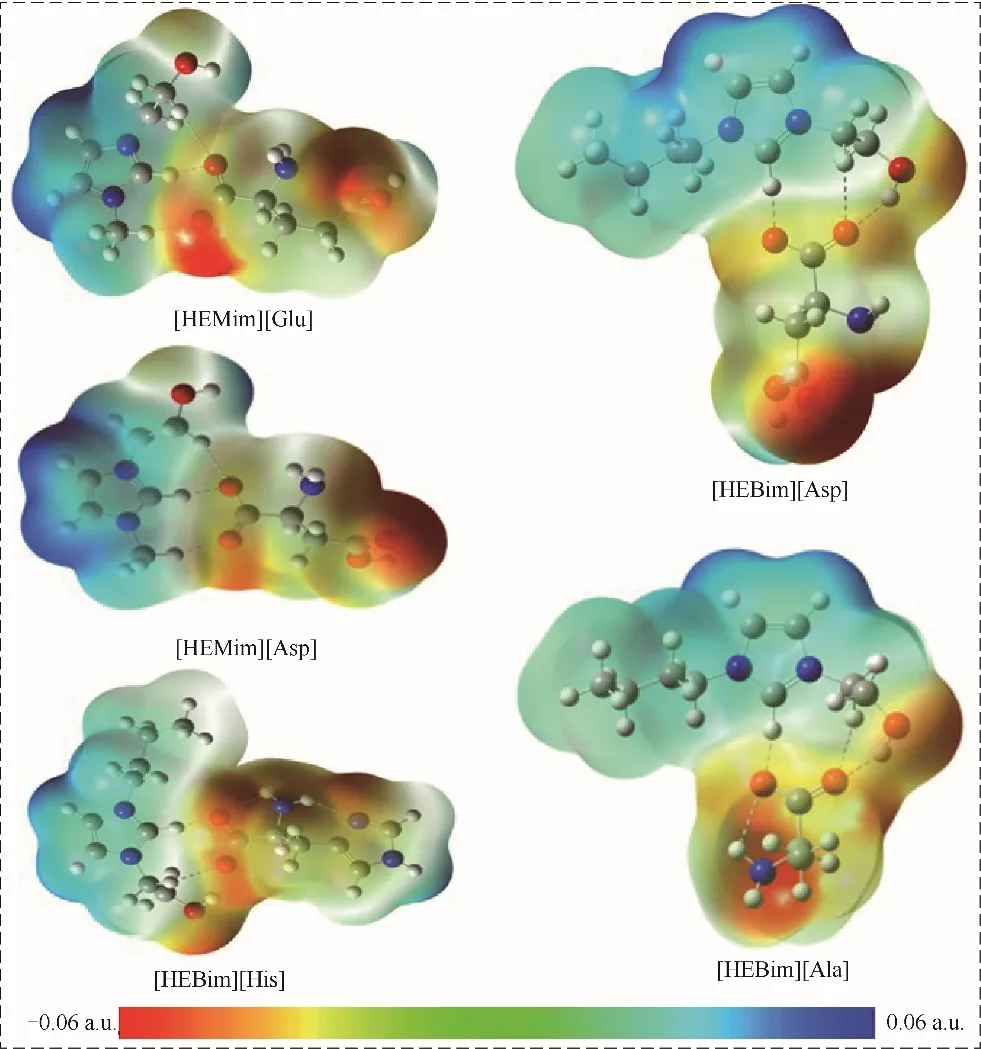
图3 在B3LYP∕6-31’++G(d,p)基组水平下得到的五种离子液体的静电势能面图Fig.3 Electrostatic potential surface of five ionic liquids at B3LYP∕6-31’++G(d,p)level
表2 给出了ILs 与气体分子可能的相互作用点的ESP、Hirshfeld和ADCH电荷。Hirshfeld电荷通常比ADCH 电荷小,ESP 的再现性差,因为它忽略了原子偶极矩。与Hirshfeld 电荷相比,ADCH 电荷能很好再现ESP,并且可以准确地预测潜在的作用位点。例如:[HEMim][Glu]的电荷分析结果显示H14 原子具有最大的正电荷,阳离子羟基中H14 原子是最有可能与NH3相互作用的原子,这与Li 等[28]和Yuan等[29]的结论相一致。[HEBim][Ala]的电荷分析得出H29 原子具有最高的正电荷,其可能是与NH3相互作用的最佳位点,而N39原子具有最高的负电荷,可能是与CO2相互作用的最佳位点。然而,H6 原子和H29 原子应该具有较高的正电荷,但静电势分析的结果却与电荷分析结果不一致,最主要的原因是H6与H29原子和O原子之间形成氢键导致静电势分析结果出现误差。

表2 在B3LYP/6-31’++G(d,p)基组下计算的离子液体的ESP、ADCH和Hirshfeld 电荷Table 2 ESP,ADCH and Hirshfeld charges for ionic liquids calculated at B3LYP/6-31’++G(d,p)level
2.3 离子液体与NH3的相互作用
在B3LYP杂化泛函6-31’++G(d,p)基组水平上对ILs与NH3的相互作用进行结构优化和频率计算,以确定ILs 吸收NH3的作用方式。图4 是优化后能量最低、最稳定的ILs与NH3相互作用的结构。可以看出,NH3主要与ILs阳离子中的羟基相互作用。通过键长与键角分析可以得出阳离子羟基中的H 原子与NH3中的N 原子形成了O—H…N 型氢键,这与前面的静电和电荷分析中潜在的作用位点相一致。表3 给出了ILs 与NH3相互作用的电子密度拓扑分析结果。结果显示:ILs 的阳离子羟基中的H 与NH3的N 形成的氢键的键长在1.780~1.796 Å 之间,ρBCP在0.043~0.045 a.u.之间,形成的氢键结合能在37.0~38.6 kJ·mol-1之间,其键长、ρBCP与∇2ρBCP均符合形成氢键的条件,且也符合ρBCP越大氢键越强的规律。从以上分析可以得出五种ILs 吸收NH3的能力为[HEBim] [His] > [HEBim] [Ala] > [HEMim] [Asp] >[HEMim][Glu] >[HEBim][Asp]。其中,[HEBim][His]吸收NH3的能力最强,其形成氢键的键长为1.7883 Å,电子密度为0.0446 a.u.,形成的氢键结合能为38.52 kJ·mol-1。

表3 在B3LYP/6-31’++G(d,p)基组水平下计算得到的五种离子液体与气体作用的键长以及电子密度性质Table 3 The bond lengths and electron density of five ionic liquids interacting with gases calculated at B3LYP/6-31’++G(d,p)level
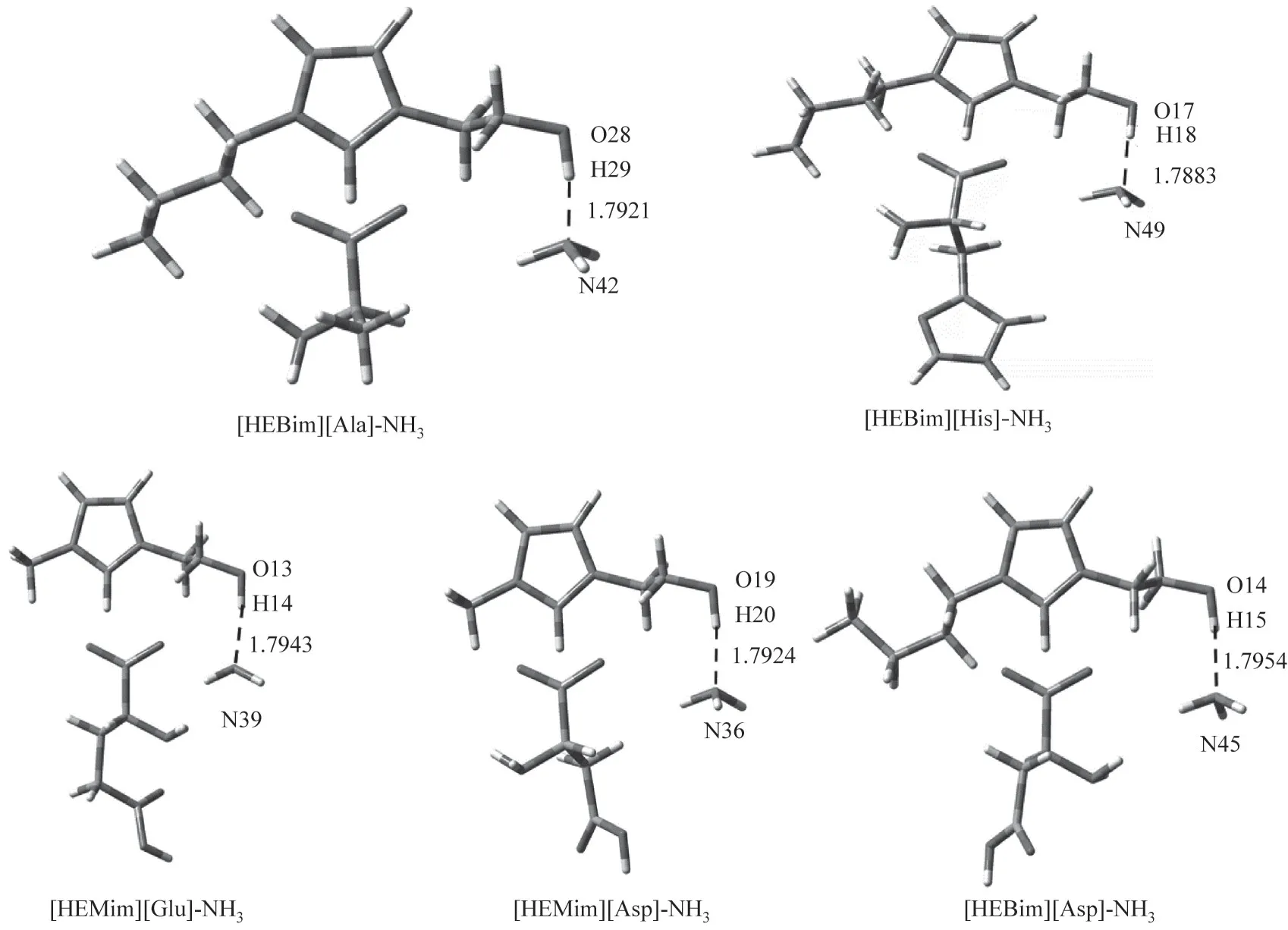
图4 在B3LYP∕6-31’++G(d,p)基组水平下优化得到的五种离子液体和氨相互作用的稳定构型Fig.4 Optimized structures of five ionic liquids and ammonia interaction at B3LYP∕6-31’++G(d,p)level
2.4 离子液体与CO2的相互作用
在B3LYP 杂化泛函6-31’++G(d,p)基组水平上进行ILs 与CO2相互作用的结构优化和频率计算。图5 是经过优化后的最稳定结构。可以看出CO2主要与ILs阴离子氨基中的N原子起作用,这也验证了上述电荷分析和ILs 静电势分析的结果。经过键长与键角分析可以看出,CO2与氨基中的N 原子形成C—N…C 型氢键,键长大都在2.8~3.2 Å 之间。从表3 可以看出ILs 中的N 原子和CO2的C 原子的ρBCP在0.007~0.015 a.u.之间,形成的氢键结合能在3.0~11.0 kJ·mol-1之间,属于较弱的氢键。根据形成氢键的强弱可以得出所设计的五种离子液体吸收CO2的强弱顺序为[HEBim][Ala]>[HEBim][His]>[HEMim][Glu]>[HEMim][Asp]>[HEBim][Asp]。其中,[HEBim][Ala]吸收CO2的能力最强,其形成的氢键的键长为2.8215 Å,ρBCP为0.0142 a.u.,形成的氢键结合能为10.15 kJ·mol-1。

图5 在B3LYP∕6-31’++G(d,p)基组水平下优化得到的五种离子液体和二氧化碳相互作用的稳定构型Fig.5 Optimized structures of five ionic liquids and carbon dioxide interaction at B3LYP∕6-31’++G(d,p)level
2.5 离子液体与NH3和CO2的相互作用
从ILs 分别与NH3和CO2的作用可以看出,虽然NH3和CO2与ILs 均是以氢键的方式进行结合的,但作用的分别是ILs 的阴离子和阳离子。考虑到氨法碳捕集中ILs 添加剂协同吸收NH3和CO2的效果,对其协同吸收时的相互影响需进一步分析。针对单独作用时优选的[HEBim][His]与[HEBim][Ala],在B3LYP 杂化泛函6-31’++G(d,p)基组水平上对其同时与NH3和CO2的相互作用进行结构优化和频率计算,其优化后的ILs与气体相互作用的结构如图6所示,结果表明:ILs同时与NH3和CO2相互作用时结构较稳定,无虚频存在。NH3主要与阳离子中的羟基形成O—H…N 型氢键,CO2则主要与阴离子中的氨基形成C—N…C 型氢键,与NH3和CO2单独与ILs 作用的结论相一致。两种ILs 与气体分子协同作用的电子密度拓扑分析如表4 所示,可以看出ILs 与NH3和CO2同时作用时的吸收能力均有不同程度的下降。例如:[HEBim][His]与NH3作用形成O17—H18…N48 型氢键,其键长为1.7886 Å、键角为169°,ρBCP为0.0445 a.u.,∇2ρBCP为0.1034 a.u.,氢键结合能为38.43 kJ·mol-1。[HEBim][His]与CO2作用形成C41—N44…C53 型氢键,其键长为2.9830 Å,键角为110°,ρBCP为0.0112 a.u.,∇2ρBCP为0.0345 a.u.,氢键结合能为7.35 kJ·mol-1。上述结果符合氢键的判据,同时也符合键长越短电子密度越大,氢键结合能越大的规律。通过分析可以看出对于吸收NH3的能力,[HEBIM][His]为38.43 kJ·mol-1,稍强于[HEBim][Ala]。而对于吸收CO2的能力,[HEBim][Ala]则稍强,为8.93 kJ·mol-1,两者在抑制氨逃逸和吸收CO2方面的差异较小。为了进一步研究ILs 协同吸收NH3和CO2时吸收能力下降的原因,在B3LYP 杂化泛函6-31’++G(d,p)基组水平上对其进行结构优化和频率计算,得到如图7 所示的光谱图。通过光谱分析可以看出:[HEBim][His]同时与NH3和CO2作用时羟基的吸收峰由3179左移到3115 cm-1,表明力常数变小,化学键变弱,使得吸收NH3的能力下降。
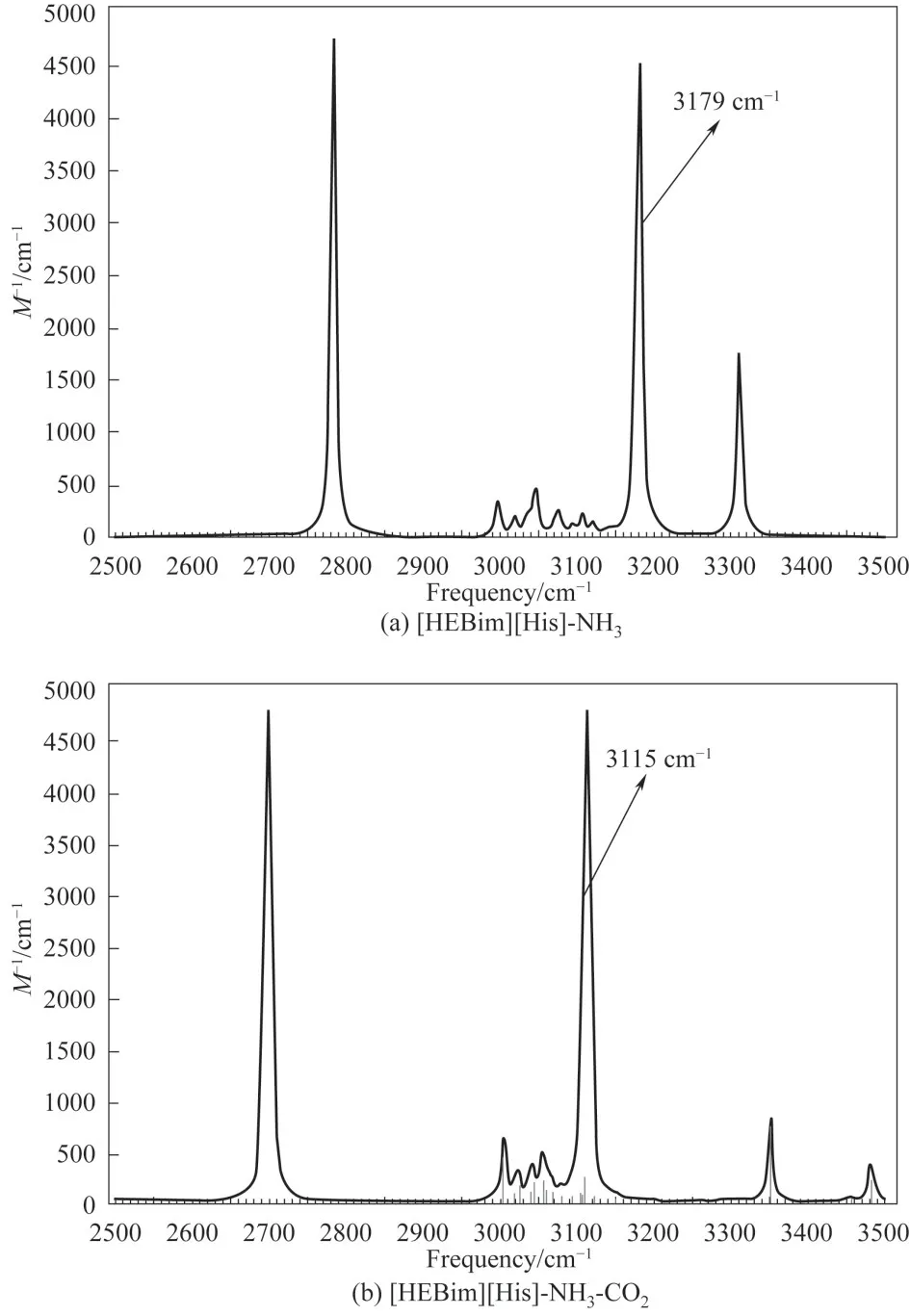
图7 优化后的离子液体与气体相互作用的光谱图Fig.7 Spectra of interaction between ILs and gases after optimization
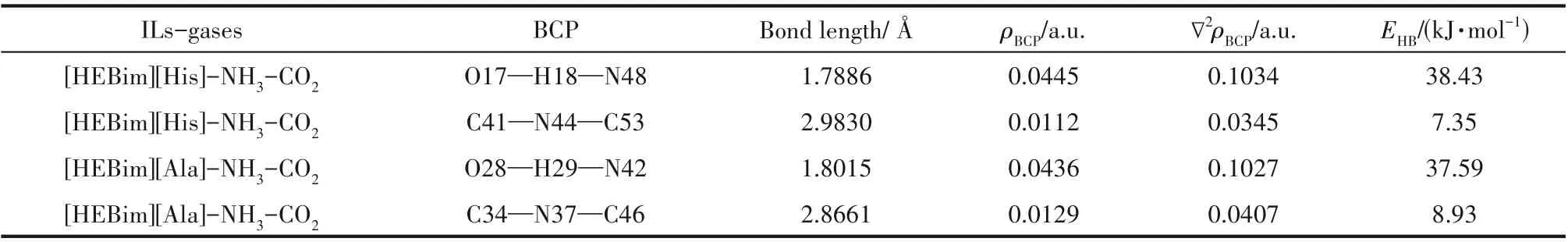
表4 在B3LYP/6-31’++G(d,p)基组水平下计算得到的两种离子液体与气体共同作用的键长、结合能以及电子密度等性质Table 4 The bond lengths,binding energies and electron densities of the two ILs interacting with gas calculated at the B3LYP/6-31++G(d,p)level
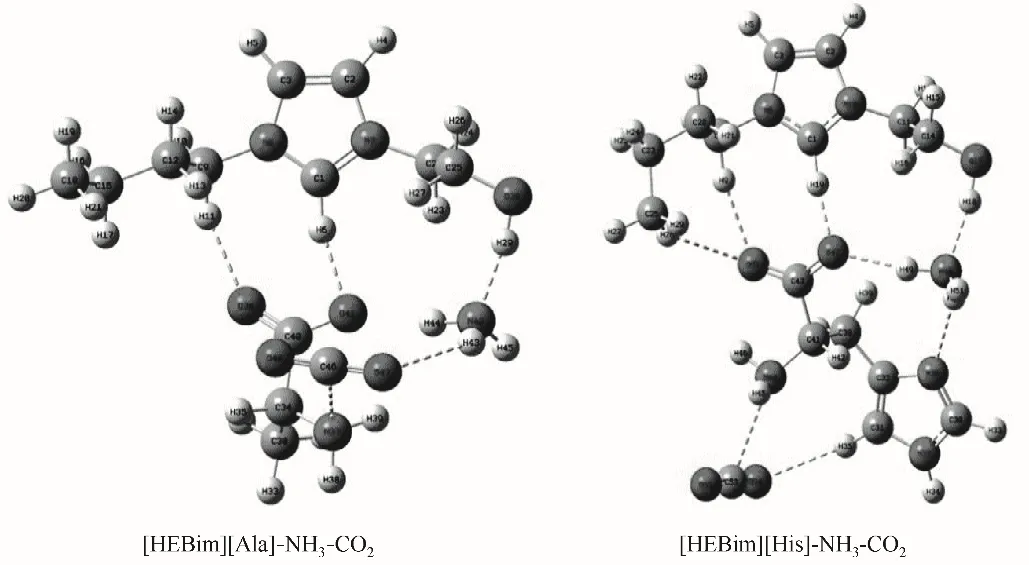
图6 在B3LYP∕6-31’++G(d,p)基组水平下优化得到的两种离子液体与气体同时作用的稳定构型Fig.6 Two stale configurations of two ILs interacting with gases obtained at B3LYP∕6-31’++G(d,p)level
为了进一步研究ILs 同时与NH3和CO2的作用实质,采用了式(5)所示的约化密度梯度(RDG)方法来研究其弱相互作用[30],这种方法可以描述包括氢键在内的不同类型分子之间的弱相互作用。
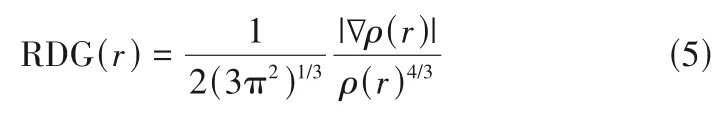
因为电子密度只能描述相互作用的强度,而相互作用类型需要由第二个Hessian 矩阵的特征值符号来确定。例如sign(λ2)=1 和sign(λ2)=-1 分别表示无键和有键的相互作用。通过图8 可以看出,在低密度和低梯度的区域内存在许多尖峰,表明ILs 与气体之间存在弱相互作用。这些尖峰可以分为三类:强吸引相互作用如氢键[大且负的sign(λ2)ρ(r)]、范德华相互作用(接近0)以及强排斥相互作用如空间位阻效应[大且正的sign(λ2)ρ(r)]。图8(a)的尖峰表示分子内和分子间的氢键。ILs 与气体相互吸引作用的sign(λ2)ρ(r)值总结在表5 中。从表中可以看出[HEBim][His]与NH3和CO2之间都形成了氢键,其中[HEBim][His]与NH3的sign(λ2)ρ(r)值达到-0.0452 a.u.,表明O17—H18…N48 之间形成了较强的氢键,与CO2的sign(λ2)ρ(r)值 为-0.0092 a.u.,表 明C41—N44…C53之间形成了较弱的氢键,符合上述电子密度拓扑分析的结论。
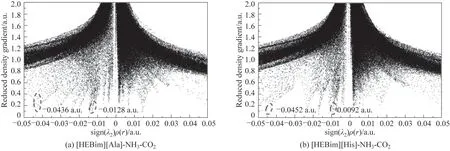
图8 通过第二个Hessian矩阵的特征值计算得出的离子液体与气体相互作用的电子密度RDG图Fig.8 Plot of RDG versus electron density multiplied by the sign of the second Hessian eigenvalue(λ2)for ILs-gas structures

表5 离子液体与气体之间的氢键的sign(λ2)ρ(r)值Table 5 sign(λ2)ρ(r)values for H-bonds between ILs and gases
3 结 论
本文针对氨法碳捕集中NH3的高挥发性和CO2吸收慢的问题,设计了五种含有羟基及氨基的功能型ILs,即[HEMim] [Glu]、[HEMim] [Asp]、[HEBim][Asp]、[HEBim][Ala]、[HEBim][His],以探究其协同吸收NH3和CO2的作用方式。通过量子化学密度泛函理论研究了ILs 之间的构型及其与气体分子之间的作用机理。相互作用能计算结果表明:[HEBim][His]的结构最为稳定,其内部形成了较多的氢键。电子密度拓扑分析表明:ILs的阳离子羟基中的H原子与NH3中的N 原子形成了O—H…N 型氢键,而CO2主要与阴离子中的氨基形成了较弱的C—C…N 型氢键。五种ILs 吸收NH3的能力为[HEBim][His]>[HEBim] [Ala] >[HEMim] [Asp] >[HEMim] [Glu] >[HEBim] [Asp],吸 收CO2的 能 力[HEBim] [Ala] >[HEBim] [His] > [HEMim] [Glu] >[HEMim] [Asp] >[HEBim][Asp]。在同时抑制NH3逃逸和促进CO2吸收性能方面,[HEBim][His]和[HEBim][Ala]均表现出了较好的效果,是比较适合的吸收剂。
猜你喜欢
河北师范大学学报(自然科学版)(2022年5期)2022-09-20
原子与分子物理学报(2022年3期)2022-03-05
中国化妆品(2022年2期)2022-03-04
波谱学杂志(2021年3期)2021-09-07
辽宁科技大学学报(2021年2期)2021-07-22
青岛大学学报(工程技术版)(2019年2期)2019-09-10
商情(2017年30期)2017-09-18
江苏农业科学(2016年1期)2017-05-17
绿色科技(2017年4期)2017-03-23
中学化学(2015年12期)2016-01-19
