
高效液相色谱法测定蒲公英提取物中10 种活性物质
2022-11-12 03:03:50李健华黄锦波
化学分析计量 2022年10期
李健华,黄锦波
[1.广东省江门生态环境监测站,广东江门 529000; 2.吉姆斯(广州)实验技术有限公司,广州 510000]
蒲公英具有清热、祛火、解毒和利尿等功效[1],可用作蔬菜、药材、饲料与保健品[2],蒲公英提取物应用范围更广[3]。蒲公英在全国各地均有种植,如安徽,东北三省,广东信宜等。不同产区的蒲公英提取物中的活性物质有一定的区别[4]。建立蒲公英提取物中活性物质含量分析方法,既可区别不同产区的蒲公英提取物,也可初步判断哪些地区的土壤或者气候条件更适合种植蒲公英,同时也能给相关的种植农户提供一定的技术参考[5-7]。
目前,测定蒲公英提取物中活性物质的方法主要有液相色谱法[8]和液相色谱-串联质谱法[9]。杨丽群[10]采用高效液相色谱法测定蒲公英提取物中的咖啡酸、绿原酸、原儿茶酸;冀新花[11]用高效液相色谱法测定蒲公英提取物中绿原酸;李盛建等[12]利用高效液相色谱-二极管阵列检测器法同时测定蒲公英提取物配方颗粒中绿原酸、咖啡酸等6 种成分的含量;原琦等[13]利用高效液相色谱-质谱法分析蒲公英中4 种有效成分。但笔者参考文献[14],采用高效液相色谱-串联质谱法测定蒲公英提取物中10 种活性物质时发现,儿茶素需要的雾化温度高,氮气消耗量大,不利于控制检测成本,而绿原酸、α-红没药醇、去甲二氢愈创木酸在正负模式下无明显响应。目前采用液相色谱法同时测定蒲公英提取物中儿茶素、红景天苷、绿原酸、阿魏酸、α-红没药醇、去甲二氢愈创木酸、甘草酸铵、甘草次酸、槲皮素、山奈酚10 种活性物质的研究还未见报道。笔者建立了高效液相色谱法测定蒲公英提取物中上述10 种活性物质的含量,该方法分析效率高,成本低,为进一步研究蒲公英的活性成分提供了一种可行的检测方法,也可以初步判断哪些地区的土壤或者气候条件更适合种植蒲公英,为相关的种植农户提供一定的技术参考。
1 实验部分
1.1 主要仪器与试剂
高效液相色谱仪:LC-1260 型,配有二极管阵列检测器(DAD)和自动进样系统,美国安捷伦科技有限公司。
电子分析天平:BSA224S-CW 型,感量为0.1 mg,德国赛多利斯科学仪器有限公司。
儿茶素、红景天苷、甘草酸铵对照品:质量分数分别为98.1%、99.7%、94.4%,批号分别为110877-202005、110818-202009、10731-202122,中国食品药品检定研究院。
α-红没药醇、去甲二氢愈创木酸对照品:质量分数分别为99%、98.0%,批号分别为CDCTGS09010039IP、CDCT-C15644200,上海安谱实验科技股份有限公司。
绿原酸、阿魏酸、甘草次酸、槲皮素、山奈酚纯度标准物质:质量分数分别为99.4%、99.8%、99.9%、99.3%、99.5%,编号分别为GBW(E) 09528、GBW(E)09518、GBW(E) 09530、GBW(E) 09526、GBW(E)09525,中国医学科学院药物研究所。
蒲公英提取物样品:市售。
实验用水为去离子水,由赛多利斯Sartorius arium®comfort Ⅱ型纯水仪制备。
1.2 溶液配制
10 种活性物质混合对照品储备液:分别称取儿茶素、红景天苷、绿原酸、阿魏酸、α-红没药醇、去甲二氢愈创木酸、甘草酸铵、甘草次酸、槲皮素、山奈酚对照品适量,用甲醇溶解并稀释至各组分质量浓度均约为1 000 μg/mL 的混合对照品储备液。
10 种活性物质系列混合标准工作溶液:精密移取10 种活性物质混合对照品储备液0.01、0.05、0.1、0.2、0.4、0.8、1.0 mL ,分别置于7 只10 mL 棕色容量瓶中,用甲醇稀释至标线,配制成各组分质量浓度均分别为1、5、10、20、40、80、100 μg/mL 的系列混合标准工作溶液。
1.3 色谱条件
色 谱 柱:SCION-C18柱(250 mm×4.6 mm,5 μm,美国瓦里安仪器有限公司);柱温箱温度:35℃;检测波长:儿茶素、去甲二氢愈创木酸、α-红没药醇为200 nm,甘草酸铵、甘草次酸为272 nm,红景天苷、绿原酸、阿魏酸为316 nm,槲皮素、山柰酚为366 nm;进样体积:10 μL;流动相:A 为甲醇,B 为0.2%甲酸水溶液;洗脱方式:梯度洗脱,洗脱程序见表1。
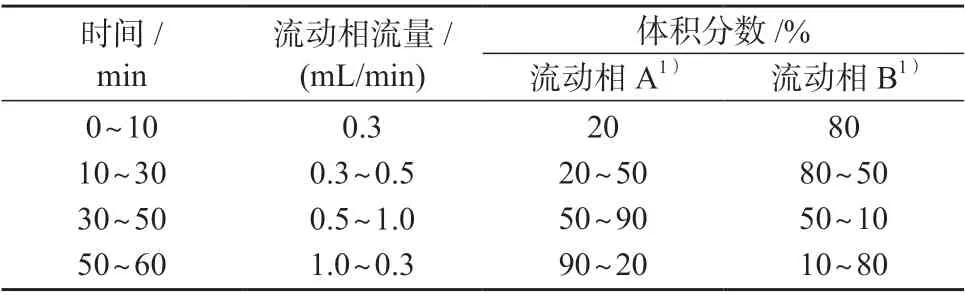
表1 梯度淋洗程序
1.4 样品处理
称取样品约1 g(精确到0.1 mg)于10 mL 棕色容量瓶中,加入2 mL 水,混匀,超声15 min,再加入5 mL 甲醇,继续超声15 min,用甲醇稀释至标线,过滤,即得样品溶液。
1.5 定量方法
取10 种活性物质系列混合标准工作溶液和样品溶液,按1.3 色谱条件进样分析,以待测组分的质量浓度为横坐标(x)、对应的色谱峰面积为纵坐标(y)绘制标准工作曲线,采用外标法进行定量。样品中活性物质的质量分数w按照式(1)计算:

式中:w——样品中活性物质的质量分数,μg/g;
ρ——样品溶液中活性物质的质量浓度,μg/mL;
V——样品溶液定容体积,mL;
m——样品质量,g。
2 结果与讨论
2.1 仪器工作条件优化
2.1.1 分析波长
采用二极管阵列检测器在190~400 nm 波长范围内对样品溶液进行扫描,结果发现,儿茶素、α-红没药醇、去二甲愈木酸在200 nm 处响应最强,甘草酸铵、甘草次酸在272 nm 处有较强的吸收,红景天苷、绿原酸、阿魏酸在316 nm 处有较强的吸收,而槲皮素、山奈酚在366 nm 处的吸收最强,因此选择儿茶素、α-红没药醇、去二甲愈木酸的分析波长为200 nm,甘草酸铵、甘草次酸的分析波长为272 nm,红景天苷、绿原酸、阿魏酸的分析波长为316 nm,槲皮素、山奈酚的分析波长为366 nm。
2.1.2 色谱柱
分别选用SCION-C18型反相色谱柱、ZORBAX Rx-SIL 型正相色谱柱和APS-2 Hypercil 型正反两用色谱柱进行试验。结果发现,使用ZORBAX Rx-SIL 型色谱柱时不出峰,使用APS-2 Hypercil 型色谱柱时仅出现1 个色谱峰,而选用SCION-C18型色谱柱时10 种活性物质出峰良好,分离度与响应值均比较理想,因此选用SCION-C18型色谱柱作为分析柱。
2.1.3 流动相配比与流量
参考文献[15],选择甲醇-0.2%甲酸水溶液为流动相。由于10 种活性物质的极性不同,需选择梯度淋洗方式进行分离。红景天苷、儿茶素、绿原酸极性相对较大,出峰时间较早,为了延长其出峰时间,在0~10 min 时,水相体积分数保持80%;阿魏酸、槲皮素与山奈酚的极性相对偏中性,出峰时间较儿茶素、绿原酸晚,故在10~30 min 时,水相体积分数由80%逐渐减小到50%;而甘草酸铵、甘草次酸、红没药醇的极性相对较弱,在30~50 min 时,水相体积分数由50%逐渐减小到10%,在50~60 min 时,水相体积分数再从10%调回80%。
一般而言,以十八烷基硅烷键合硅胶为填充剂的色谱柱(简称C18柱)作为分析柱时,流动相的流量越大,目标色谱峰保留时间越小[16]。当流动相流量为1 mL/min 时,红景天苷色谱峰与儿茶素色谱峰发生重叠,分离度小于0.5。为了提高色谱峰分离度,在0~10 min 时,流动相流量为0.3 mL/min,在10~30 min 时流量为0.5 mL/min。由于30 min 后目标峰仅余2 个,因此在30~50 min 流动相流量改为1 mL/min,以缩短分析时间,在50~60 min 时,流量则改回0.3 mL/min。
在优化的仪器工作条件下,10 种活性物质混合标准溶液和加标样品溶液的色谱图分别如图1 和图2 所示。
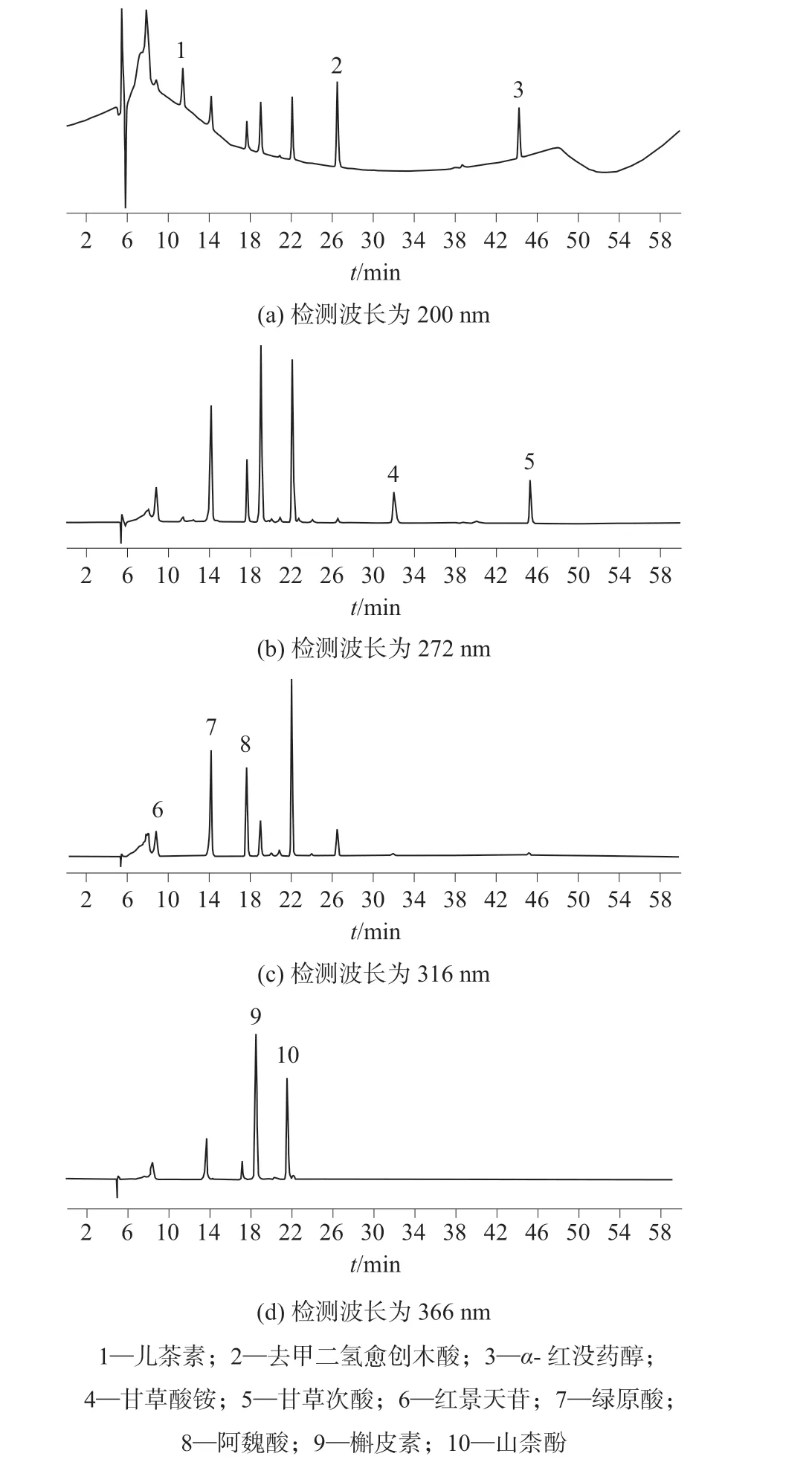
图1 10 种活性物质混合标准溶液色谱图
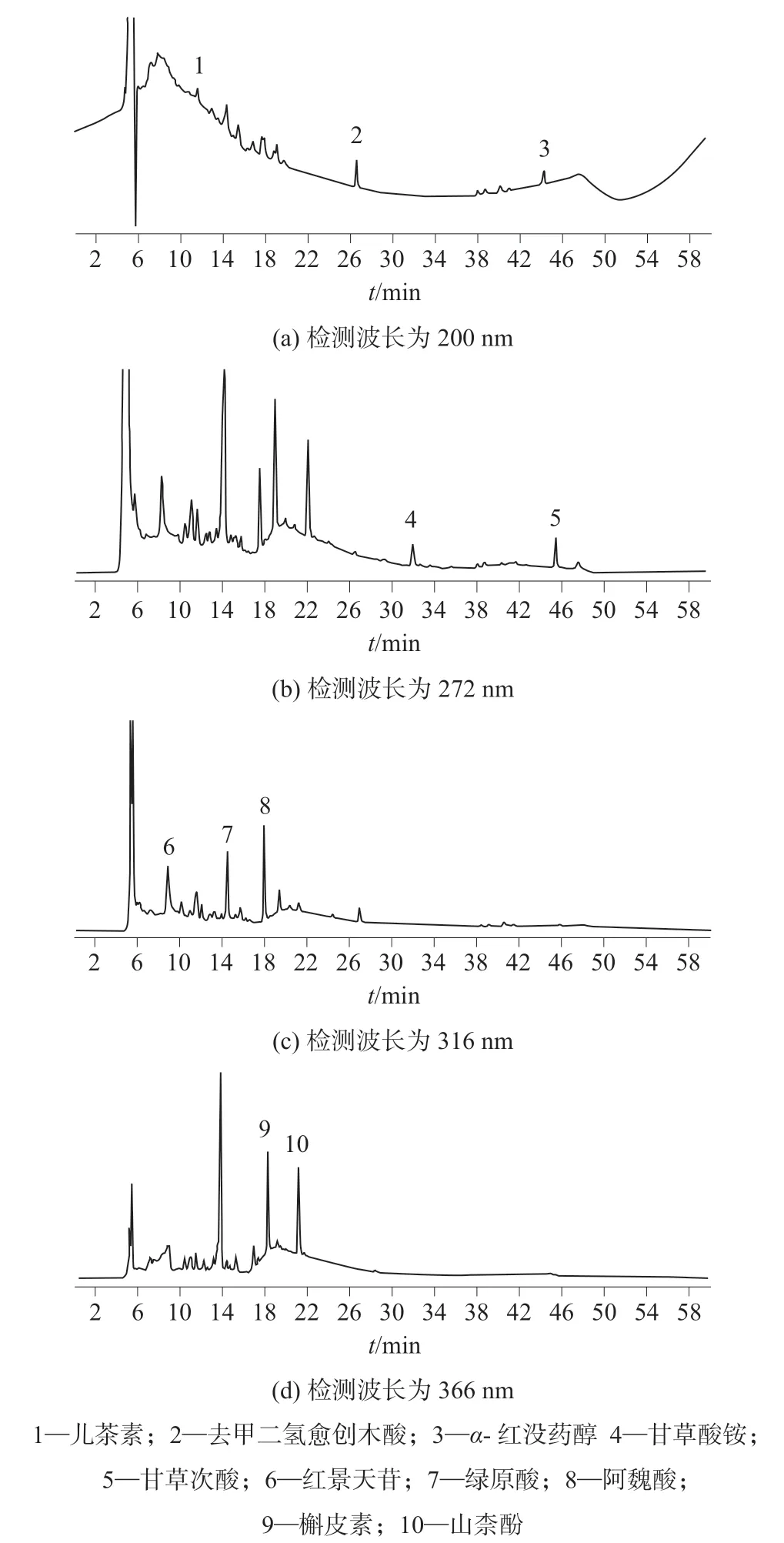
图2 加标样品溶液的色谱图
2.2 线性方程与检出限
在1.3 色谱条件下,分别测定10 种活性物质系列混合标准工作溶液,以各组分质量浓度为横坐标(x)、色谱峰面积为纵坐标(y)绘制标准工作曲线,计算线性方程和相关系数。
取10 种活性物质的质量浓度均为20 μg/mL的混合标准溶液,用甲醇逐级稀释,在1.3 色谱条件下进行测定,记录色谱峰面积,以3 倍信噪比对应的质量浓度作为待测物质的检出限。
10 种活性物质的质量浓度线性范围、线性方程、相关系数及检出限见表2。
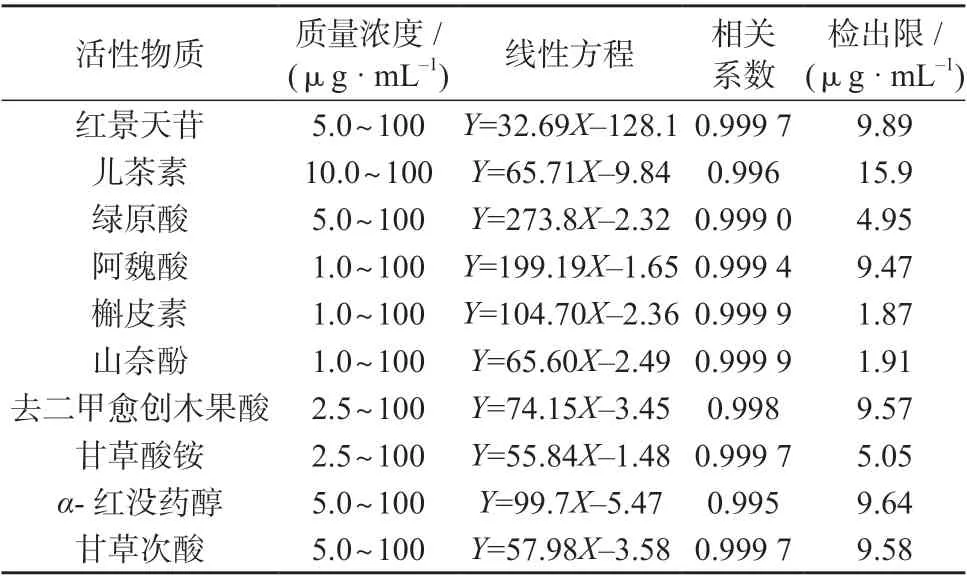
表2 10 种活性物质的质量浓度线性范围、线性方程、相关系数及检出限
2.3 加标回收与精密度试验
取蒲公英提取物样品,测定其中10 种活性物质的含量。分别向样品中添加0.16、0.32、0.8 mL的10 种活性物质混合对照品储备液,按1.4 方法每种加标浓度水平平行处理6 份样品溶液,在1.3 色谱条件下分别进行测定,计算各待测物质的加标回收率和测定结果的相对标准偏差,结果见表3。由表3 可知,10 种活性物质的加标回收率为86.2%~104.3%,测定结果的相对标准偏差为0.2%~4.8%。表明该方法具有良好的准确度和精密度,可用于检测蒲公英提取物中的10 种活性物质。
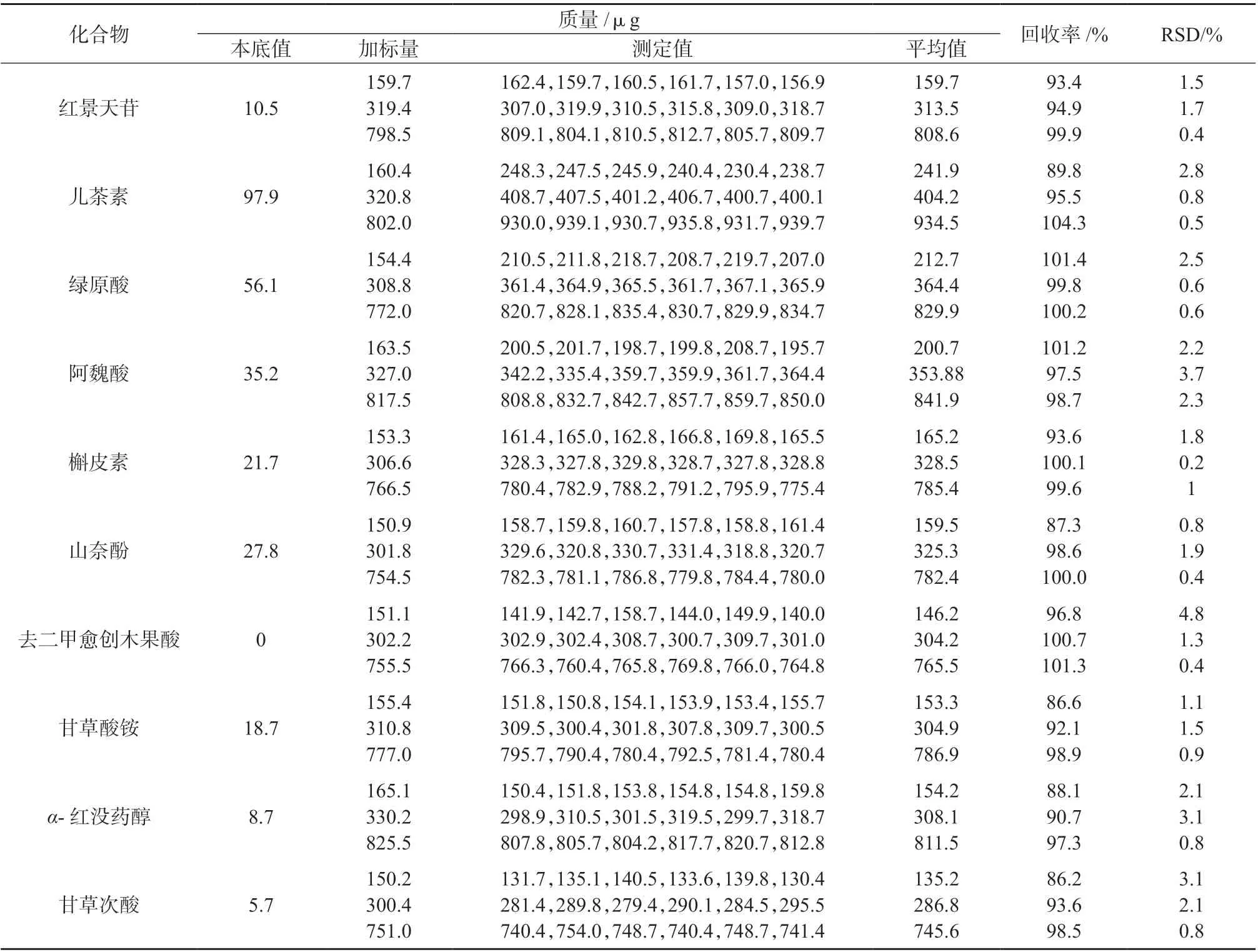
表3 加标回收与精密度试验结果
3 结语
建立了高效液相色谱法同时测定蒲公英提取物中10 种活性物质的含量,采取梯度淋洗方式进行分离,利用二极管阵列检测器检测,提高了分析效率,降低了分析成本,方法精密度、准确度高。
猜你喜欢
今日农业(2022年14期)2022-09-15 01:45:16
食品安全导刊(2021年20期)2021-08-30 06:39:40
天然产物研究与开发(2019年1期)2019-03-01 05:41:08
中成药(2018年11期)2018-11-24 02:56:46
知识经济·中国直销(2017年12期)2018-01-03 08:21:20
中成药(2017年10期)2017-11-16 00:50:42
国际心血管病杂志(2015年5期)2015-02-27 12:11:37
中国药业(2014年24期)2014-05-26 09:00:11
中国海洋大学学报(自然科学版)(2014年12期)2014-02-28 12:22:07
茶叶通讯(2014年2期)2014-02-27 07:55:38
