
14例甲基丙二酸血症患儿临床表型、基因变异分析及治疗效果评价
2022-11-08 12:20:54穆文娟郝丽婷董勤杨建平
中国生育健康杂志 2022年6期
穆文娟 郝丽婷 董勤 杨建平
甲基丙二酸血症(methylmalonic aciduria,MMA)也称甲基丙二酸尿症(methylmalonic aciduria),是国内先天性有机酸代谢异常中最常见的疾病[1]。根据酶缺陷的类型分为甲基丙二酸辅酶A变位酶缺陷(methylmalonyl coenzyme A mutase)及其辅酶维生素B12(钴胺素)代谢障碍两大类,迄今共发现10个亚型,其中钴胺素代谢障碍有cb1A、cb1B、cb1C、cb1D、cb1F及cb1H6个亚型;Mut、cb1A、cb1B及cb1H缺陷型仅表现为MMA,称为单纯性MMA;cb1C、cb1D和cb1F缺陷型则表现为MMA伴同型半胱氨酸血症,故称为合并型MMA;除cb1X型为X连锁遗传,其余9种亚型均为常染色体隐形遗传[1]。MMA患儿临床表现个体差异较大,发病年龄越早病情越重,重者于新生儿期发病死亡,轻者可晚至成年发病[2]。MMA常因发热、饥饿、高蛋白饮食、感染等诱发代谢性酸中毒急性发作,若不能及时诊治,猝死率极高[3]。本文回顾性分析了14例MMA症患儿的临床资料、辅助检查结果及预后随访,为本病的临床诊断和遗传咨询提供一定的依据。
对象与方法
1.研究对象:选取2016年10月~2020年4月在山西省儿童医院经基因检测确诊为MMA的14例患儿作为研究对象,其中男9例,女5例。14例患儿中9例(64.3%)通过液相串联质谱法新生儿筛查发现,5例(35.7%)为发病后临床诊断,发病年龄16 h~7岁不等,14例患儿均接受了基因突变检测。
2.血浆液相色谱串联质谱检测:14例确诊患儿均经末梢血采血,采用液相串联质谱技术(Waters Quattro API 串联质谱仪,试剂购自PerkinEImer),进行多种氨基酸和酰基肉碱分析;检测方法参考文献[4]。其中血游离肉碱、丙酰肉碱(propionylcarnitine,C3)、蛋氨酸及C3与乙酰肉碱(acetylcarnitine,C2)比值(C3/C2)的正常参考值分别为10~62 umol/L、0.32~4.66 umol/L、4.3~36.3 umol/L、0.04~0.42(本切值为本实验室以10 000例新生儿干血斑的串联质谱检测结果为资料,录入氨基酸、有机酸、 脂肪酸相应浓度值,建立数据库所得,参考文献[5~6])。初筛血C3及C3/C2比值增高立即召回重新采集血样进行复查,同时进行尿液串联质谱测。
3.尿液气相色谱质谱检测:采患儿尿液,制成尿滤纸片送检,采用气相串联质谱技术,应用QP2010型气相色谱质谱仪(日本岛津公司)分析尿液中有机酸。其中尿甲基丙二酸和甲基枸橼酸水平,正常参考值分别为0.0~4.0 umol/L、0~1.0 umol/L,(切值参考厂家说明书,参考文献[6])明显增高者,结合血串联质谱分析,临床诊断为MMA,立即进行基因突变分析。
4.基因突变分析:在家长知情同意下,留取14例患儿及其父母静脉血4 mL(EDTA抗凝),使用BloodGen Midi Kit(CWBIO,China)提取外周血白细胞DNA。采用IDT公司xGen© Exome Research Panel v1.0捕获探针与gDNA文库序列进行液体杂交,将目标区域DNA片段进行富集,构建全外显子文库。通过illumina公司NovaSeq 6000系列测序仪进行高通量测序,目标序列测序覆盖度不低于99%;对于可疑致病突变位点,利用Sanger测序对患儿及其父母样本的该位点进行验证分析。
5.其他辅助检查:包括血常规、血糖、血氨、血乳酸、血同型半胱氨酸、肝肾功能、心肌酶谱、血气分析、 头颅MRI等。
6.病例的诊疗及随访:(1)诊断依据。诊断依据参照《临床遗传代谢病》[1]。(2)治疗。 MMA患者以减少甲基丙二酸及其旁路代谢产物的生成和加速其清除为治疗原则。具体治疗参照《从病例开始学习遗传代谢病》[7]。(3)随访。对确诊患儿进行电话或门诊随访,每1~3个月随访1次,稳定患者每半年~1年随访1次,随访内容包括体格发育及智力评估、血、尿串联质谱、血同型半胱氨酸,血系列、肝肾功能、电解质、血氨及血乳酸等。其中智能发育评估采用儿童心理行为发育量表(包括适应性、精细动作、大动作、语言和个人-社交5个能区)测定发育商(developmentquotient,DQ)[8]。
7.评价指标:血中C3,C3/C2下降情况;尿中甲基丙二酸和甲基枸橼酸下降情况;血同型半胱氨酸水平;患者转归。
8.统计学处理:采用SPSS 22.0统计学软件进行数据分析,定性资料采用频数和构成比(%)描述,行χ2检验;定量资料以均数±标准差表示,采用t检验,以P<0.05为差异有统计学意义。
结果
1. 山西省MMA患病情况:山西省新生儿疾病筛查中心在2016年至2020年间,由所管辖6个市(太原地区、临汾地区、吕梁地区、忻州地区、晋中地区、阳泉地区)的173 117例活产儿中发现9例MMA,故山西部分地区MMA患病率约为1/19 235(9/173 117)。
2. 一般临床资料:本研究中9例患儿由新生儿期确诊,4例患儿在无症状时开始治疗,随访至 26~59月龄,发育基本正常,5例患儿发病后门诊就诊,已有不同程度的发育落后,14例MMA患儿,男9例,女5例,其中2例(例1,例7)确诊时已死亡,1例(例13)确诊后未正规治疗,在8月龄时死亡,4例确诊时无明显临床症状。除3例死亡病例,11例患儿均行头颅MRI,其中2例未见明显异常,4例双侧基底节对称性病变,3例脑白质病变,3例重度脑积水,2例脑外间隙增宽。2例患儿曾有异常家族史,1例为母亲不明原因流产或死胎史,1例为同胞不明原因新生儿早期死亡。患儿一般临床资料,见表1。
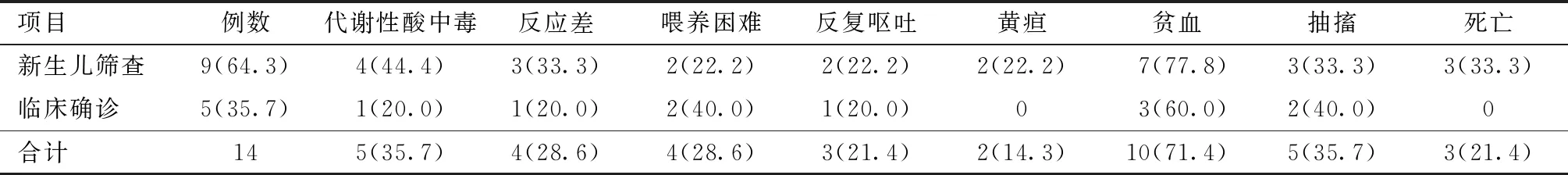
表1 14例MMA患儿的一般临床资料 [例(%)]
3.随访患者治疗前后临床表现变化:除3例死亡病例,余11例MMA患儿随访过程中4例生长发育正常(例4,例6,例9,例11),智能发育落后7例中有2例体格生长发育迟缓(例4,例10)。2例发生严重脑积水(例10,例13),1例有小头畸形(例2)。
4.血同型半胱氨酸及血红蛋白:8例合并型患者中,例1于治疗前死亡,余7例患儿治疗前后血同型半胱氨酸水平见表2。另7位合并有不同程度的贫血,予维生素B12等治疗1月后均痊愈。
5. C3、C3/C2及尿甲基丙二酸、甲基枸橼酸:7例合并型患儿血C3、C3/C2及尿甲基丙二酸、甲基枸橼酸治疗前后水平比较,差异均有统计学意义,见表2。

表2 7例合并型MMA患儿治疗前后生化检测结果比较
6例单纯型患儿中,例7于治疗前死亡,余5例患儿血C3、C3/C2及尿甲基丙二酸、甲基枸橼酸治疗前后水平比较,差异均有统计学意义,见表3。

表3 5例单纯型MMA患儿治疗前后生化检测结果比较
6. 基因突变分析:14例患儿均进行了基因突变分析(见表4);以MUT基因突变为主(48.1%,13/27),其次为MMACHC基因型(37.0%,10/27)。其中MUT基因突变6例,符合MUT型,MUT基因与MMAA基因复合杂合突变1例,7例共检出12种MUT基因突变类型,其中错义突变8种,分别为c.1880A>G,c.1280G>A,c.323G>A,c.2080C>T,c.1286A>G,c.914T>C,c.428A>G,c.2032C>G;无义突变1种,c.349G>T;经典剪切位点突变2种,分别为c.1560+1G>A、c.1677-1G>A;剪切位点突变1种,c.1084-10A>G。携带c.1560+1G>A位点的例7已于确诊时发病死亡。例11为c.1880A>G和c.1280G>A的复合杂合突变,血、尿各生化指标均可控制在正常范围,生长发育基本正常;例9为c.1084-10A>G剪切位点突变,患儿未治疗的情况下,血酰基肉碱、尿有机酸等生化指标控制在正常参考范围内,颅脑MRI未见明显异常改变,目前智能、运动、认知、语言均发育正常,例3为MMAA和MUT的复合杂合变异,分别为剪切位点突变c.819+25A>G和错义突变c.428A>G,无明显临床症状,但血C3、C3/C2及尿甲基丙二酸、甲基枸橼酸持续轻度增高。MMACHC基因突变5例,符合cblC型,共计4种MMACHC基因突变类型,其中错义突变1种,c.482G>A,无义突变2种,分别为c.217C>T、c.609G>A,移码突变1种,为c.567dup;最常见的MMACHC基因突变为c.609G>A(2例纯合突变,3例复合杂合突变),其频率占所有突变位点的7/10,例1为c.609G>A和c.482G>A的复合杂合突变,已于新生儿早期发病死亡;例13为c.609G>A与c.567dupT的复合杂合突变,在8月龄时死亡,例10、例12、例13与例14均在随访过程中出现严重的生长发育及智力的落后。MMAA基因突变1例,例4患儿MMAA基因复合杂合变异,符合cblA型,发现两种突变类型,均为错义突变,分别为c.1003A>T,c.1076G>A,只表现为血C3,C3/C2的轻度增高,未治疗的情况下,一般情况良好,无任何临床表现,CD320基因突变1例(例5)为CD320的单杂合变异,为错义突变,c.446C>T,例5因中度贫血及抽搐入院,给予B12治疗后该患儿贫血、及高同型半胱氨酸迅速恢复正常,血串联质谱C3,C3/C2水平与尿液甲基丙二酸水平降至正常。

表4 14例MMA患儿基因突变检测结果
讨论
1.MMA的患病率:MMA患病率在不同国家有很大差异,美国患病率为1.3/10万;德国为0.4/10万;日本为2/10万;中国台湾省为1.2/10万[7];在中国,MMA患病率亦有明显的地区差异性,上海地区为3/10万[1];浙江省为1.5/10万[9],山东省济南市于2011至2014年,筛查了 35 291名新生儿,发现9例MMA,患病率高达1∶3 920[10];江苏省徐州市于2015至2017年,筛查了236 368名新生儿,发现14例MMA,患病率为1∶16 883[11];河南省新乡市于2015至2018年,筛查了50 112名新生儿,发现8例MMA,患病率约1∶6 264[12]。山西省新生儿疾病筛查中心在2016年至2020年间,由所管辖六个市(太原地区、临汾地区、吕梁地区、忻州地区、晋中地区、阳泉地区)的173 117例活产儿中发现9例MMA,故推测山西部分地区MMA患病率约为1/19 235(9/173 117),证明了MMA患病的地区差异性。
2.MMA临床表现的多样性:MMA可造成多脏器损伤,尤其是神经系统损伤。患儿临床表现个体差异较大,发病年龄越早病情越重[1]。患儿常因发热、饥饿、高蛋白饮食、感染等诱发“代谢危象”[13]。若不能及时诊治,猝死率极高。本文患儿中例1于生后即因“代谢紊乱”入住于本院NICU病房,14 d死亡,例7确诊时已死亡,例13于4月龄时于一次“发热”后死亡。而平稳期患儿实验室检查常见代谢性酸中毒、巨幼细胞性贫血和血小板减少等,本文患儿中曾发生代谢性酸中毒5例,贫血10例,血小板减少2例。MMA的神经系统损伤以脑损伤最为显著,临床主要表现为脑神经结构、脑神经发育及脑功能损伤等方面[14],MRI上主要表现以苍白球及基底节区的改变为主。本文11例存活患儿头颅MRI中,2例未见明显异常,4例双侧基底节对称性病变,3例脑白质病变,3例重度脑积水,2例脑外间隙增宽;临床表现方面,1例出现了小头畸形,7例有不同程度的智力发育落后。国外Horster等[15]报道称273例Mut型患者中28%并发慢性肾衰竭;Cosson等[16]的报道中47%存在慢性肾功能损害;另外还有并发急慢性胰腺炎及心肌病的报道[17]。本研究14例患儿中,未发现合并慢性肾衰、心肌病及胰腺炎者。
3.MMA的基因型分析:
(1)MMA的不同基因型。
MMA根据酶缺陷的类型分为甲基丙二酸辅酶A变位酶缺陷(methylmalonyl coenzyme A mutase)及其辅酶维生素B12(钴胺素)代谢障碍两大类,迄今为止,共发现10个亚型[1]。本研究确诊MMA的14例患儿中,以MUT基因突变为主(48.1%,13/27),其次为MMACHC基因型(37.0%,10/27),与国内其他研究结果不一致,可能与病例数较少有关。
(2)不同基因型对治疗的反应不同。
MMA临床疗效及预后取决于患儿基因型、发现早晚与长期规范治疗等方面[18]。MUT基因定位于常染色体6p12.3,包含13个外显子,编码MCM蛋白。MCM酶缺陷分为酶活性完全丧失(MUT0型)或部分丧失(MUT-型)。MUT0型大多在新生儿期出现急性代谢性脑病表现,病死率高,预后差,MUT-型,VitB治疗效果好于MUT0型。迄今已报道的该基因突变类型280余种,多数为错义突变。国内最常见的突变位点是c.729_730insTT,其次是c.323G>A和C.1106G>A[19]。本研究发现有1例携带c.323G>A突变,未发现其余两种位点突变,可能与病例数较少有关。c.1880A>G突变造成11号外显子第627位组氨酸变为精氨酸,王斐等[14]报道称携带该突变的患者为早发型MMA,表现为呕吐、嗜睡、抽搐等严重神经系统表现,而王秀利等[20]研究中携带该位点杂合突变的患儿为轻度的MUT-型;本研究中例11患儿也携带该位点,患儿经新生儿筛查确诊,后正规治疗随访,血、尿各生化指标均可控制在正常范围,生长发育基本正常,与王秀利等[20]报道相符,因此关于该突变的基因型和表型的关系还需更多病例研究证实。另本研究中发现1例(例9)良性mut-型,患儿未治疗的情况下,血酰基肉碱、尿有机酸等生化指标控制在正常参考范围内,颅脑MRI未见明显异常改变,目前智能、运动、认知、语言均发育正常,考虑可能c.1084-10A>G基因型对酶活性的损害影响不大,但国外报道有的患者持续无症状,成年后才发病[21],故本文患者长期预后及干预时机还有待进一步随访、观察。
MMACHC基因位于常染色体lp34,编码细胞浆内的cblC蛋白,在胞浆内起催化CNCbl还原脱氰和合成腺苷钴胺素及甲基钴胺素的作用,MMACHC基因突变导致早发型(cblCI型)和晚发型(cblCⅡ型)两种表型[1]。Morel等[22]报道欧美国家cblC型MMA患者最常见的突变类型是c.271dupA,约占40%,该位点的纯合或复合杂合突变发病早,病情重,预后差;而中国的常见突变位点是c.609G>A,亦多为早发型[3],病情亦较重,预后较差。本研究中5例cblC型患者,均携带c.609G>A突变,且2例(例10、例14)为该位点的纯合突变,2例(例1、例13)均在婴儿期死亡,例10、例12、例13与例14均在随访过程中出现严重的生长发育及智力的落后,例10、例13伴有重度脑积水,与文献报道一致;另据Lerner-Ellis等的报道[23]称c.567dupT的位点突变造成4号外显子第190位异亮氨酸开始发生移码突变,对所编码蛋白产生较大的影响,故携带者也多为早发型,且症状重、预后差,本研究中例13为c.609G>A与c.567dupT的复合杂合突变,已在婴儿期死亡,与文献报道相符。Liu等[3]曾报道MMACHC基因的c.482G>A 突变频率为7%,也是国内常见的突变之一,该突变造成cblC蛋白第161位氨基酸由精氨酸变为谷氨酰胺,对蛋白质功能的影响较小,与晚发型MMA有一定相关性,且临床表现较轻。王秀利等[20]的研究中有3例患者携带c.482G>A杂合突变,至截稿之日尚未发病,且随访过程中生长发育正常,而本研究中例1为c.609G>A和c.482G>A的复合杂合突变,在新生儿早期死亡,考虑基因位点c.609G>A对蛋白质功能影响较c.482G>A大。
(3)其他基因型对治疗的反应。
本研究中例5因中度贫血及抽搐入院,入院后经血尿串联质谱考虑为MMA,基因检测检出CD320的单杂合突变,根据文献报道[19]该位点通过影响VitB12通过细胞膜及线粒体膜的转运障碍而致病,为常染色体隐性遗传,而该患儿应只为携带者不发病,但予B12治疗后该患儿贫血、及高同型半胱氨酸迅速恢复正常,血串联质谱C3,C3/C2水平与尿液甲基丙二酸水平降至正常,考虑治疗有效,故临床应诊断,与文献不符,因此关于该突变的基因型和表型还有待于进一步研究,建议该患儿行全基因组测序。本文中例4患儿检出了MMAA基因上例3c.1003A>T/c.1076G>A的复合杂合突变,只表现为血C3,C3/C2的轻度增高,未治疗的情况下,一般情况良好,无任何临床表现,例3患儿为MMAA和MUT的复合杂合变异,该患儿亦无明显临床症状,但血C3,C3/C2及尿甲基丙二酸、甲基枸橼酸持续轻度增高,考虑可能为两位点均部分轻微影响酶活性而未导致临床症状。
4.小结:综上所述,MMA病死率较高,预后较差,新生儿筛查可以对本病早发现,早治疗,减少不可逆转的并发症。本研究中9例患儿由新生儿期确诊,4例患儿在无症状时开始治疗,随访至 26~59月龄,发育基本正常,但亦有5例患儿因家长未重视新生儿筛查或所在地区未开展血浆串联质谱技术进行新生儿筛查,发病后就诊时,才确诊本病开始治疗,造成了不同程度的发育落后,故应重视新生儿筛查工作。随着医疗水平的进歩,目前国内多个省份将血浆串联质谱技术应用于常规新生儿筛查,这使本病的早期诊断率极大提高,减少了MMA婴幼儿的病死率及致残率。另由于本病妊娠有再发风险性,应重视产前诊断,对于有先证者的家庭,再次怀孕时应进行产前诊断,以指导优生优育。
猜你喜欢
故事作文·低年级(2023年11期)2023-12-05 06:39:56
故事作文·低年级(2023年12期)2023-03-24 14:16:52
种子(2021年3期)2021-04-12 01:42:22
探索科学(学术版)(2020年4期)2021-01-18 02:28:56
中国环境监察(2016年7期)2016-10-23 05:36:30
中国现当代社会文化访谈录(2016年0期)2016-09-26 08:46:23
山东化工(2016年4期)2016-09-05 12:30:54
外语教学理论与实践(2016年1期)2016-06-11 05:51:48
灾害医学与救援(电子版)(2016年2期)2016-03-11 20:18:04
医学研究杂志(2015年3期)2015-06-10 06:41:52
