
锕系金属(铀和锔)内嵌硼球烯的理论研究
2022-11-07 10:01张乃心王聪芝赵玉宝石伟群
核化学与放射化学 2022年5期
张乃心,王聪芝,赵玉宝,石伟群
1.南华大学 化学化工学院,湖南 衡阳 421001;2.中国科学院 高能物理研究所 核能放射化学实验室,北京 100049
在C60[1]被发现后不久,第一例金属内嵌富勒烯La@C60[2]被报道。随后,一系列金属内嵌富勒烯相继被研究,包括碱金属、碱土金属、过渡金属、镧系元素和锕系元素等[3-11]。
硼是元素周期表中碳的相邻元素,具有独特的缺电子特性。与碳相似,硼团簇是否可以形成球形空心结构引起了相关研究者的极大兴趣。2007年,Szwacki等[12]通过理论研究预测B80具有笼状结构,但随后Zhao等[13]研究发现核壳结构B80的能量低于空心笼结构。此后,人们对各种各样的硼团簇进行了大量的理论和实验研究[13-22]。在过去的几十年中,借助于光电子能谱和先进理论计算,硼团簇的种类和结构被不断丰富。2014年,美国布朗大学王来生和国内合作者[23]首次通过光电子能谱观测到笼状全硼富勒烯(硼球烯,D2dB4-0/0,borospherene)。硼球烯的发现,标志着硼团簇由二维结构演变为三维结构,这是硼化学史上的重大突破。随后,他们又发现了一系列不同尺寸的硼球烯[24]。在2015年,他们通过光电子能谱和理论计算对B3-9进行了系统地研究,发现这是一类新型的手性硼球烯[25]。这些硼球烯遵循硼团簇中普遍存在的σ和π双离域键合模式[25-27]。
与富勒烯相似,硼球烯也可以通过掺杂金属原子进行化学修饰和功能化。例如,2015年首次报道的碱土金属掺杂硼球烯(MB40,M=Be、Mg、Ca和Sr),密度泛函理论预测Ca@B40和Sr@B40的最稳定结构为内嵌硼球烯结构[28]。此后相关理论研究表明,过渡金属和镧系元素也可以掺杂到硼球烯中。例如,对镧系元素掺杂的硼团簇M@Bn(M=Eu、Gd,n=38、40)的理论计算预测了三个内嵌硼球烯Gd@B38、Eu@B40和Gd@B40,这是第一个基于镧系元素的金属内嵌硼球烯的研究工作[29]。另外,这些金属掺杂硼球烯一般比较稳定,具有较高的形成能和较大的最高占据轨道(HOMO)-最低空轨道(LUMO)能隙[29]。除了碱土金属、过渡金属和镧系元素外,锕系元素也可掺杂于硼球烯形成稳定的锕系金属内嵌硼球烯。2018年报道的铀内嵌硼球烯U@B40[30]是第一例锕系掺杂硼球烯。理论研究发现,U@B40具有闭壳层单重态的电子结构,并且符合32电子规则。随后一系列锕系金属掺杂的硼球烯被报道,例如AnBn(An=U、Th,n=36、38和40)[31],其 中U@B36(C2h)和Th@B38(D2h)具有几乎完美的内嵌硼球烯结构,而且稳定性很高。对于已报道锕系金属掺杂硼团簇AnB12(An=Th—Cm)[32]和AnB24(An=Th—Cm)[33],由于硼原子数相对较少,笼状结构不是其最稳定结构。最近,本课题组通过系统的理论计算研究了一系列手性锕系元素(锕和钍)内嵌硼球烯[34]。
尽管如此,目前锕系金属掺杂硼球烯方面的研究还相对较少。考虑到硼球烯种类丰富,而且尺寸各异,若将不同的锕系金属掺杂到不同尺寸的硼球烯中,有望得到稳定的锕系内嵌硼球烯。在本工作中,基于硼球烯B-39,拟采用密度泛函理论(DFT)方法,系统地探讨一系列铀和锔内嵌硼球烯[An@B39]n+(An=U,n=3;An=Cm,n=2),其中U和Cm考虑了它们在固体中常见的氧化态(+4和+3),研究结果将有助于深入理解金属掺杂硼球烯体系的结构和性质,同时可为新型稳定硼球烯的设计和开发提供理论基础。
1 计算方法
对于所有[An@B39]n+体系,使用Gaussian 16软件包进行结构优化[35]。采用DFT理论方法中的PBE0[36]杂化密度泛函进行几何优化。对于锕系金属原子(U和Cm),使用准相对论小核赝势ECP60MWB以及相应的ECP60MWB-SEG基组[37-38]。对于硼原子,采用6-311+G*基组。已有研究表明,在PBE0/6-311+G*/RECP理论水平下研究硼团簇体系能够得到合理的结果[31,39-40]。为了验证[An@B39]n+优化所得结构为势能面上的极小值,在PBE0/6-311+G*/RECP理论水平下进行了谐振频率计算,并且在相同的理论水平下考虑了所有结构的零点能(ZPE)校正。使用Multiwfn 3.7软件[41],采用适应性自然密度划分(Ad NDP)[42]和分子中原子的量子理论(QTAIM)方法[43]分析了[An@B39]n+的化学键。分子轨道(MO)的成分分析[44]、Hirshfeld布居[45]和Voronoi变形密度布居(VDD)[46]分析也是通过Multiwfn 3.7软件[41]分析得到。
2 结果与讨论
2.1 [An@B39]n+的几何结构
对于每个[An@B39]n+体系,以文献[47]报道的[Ca@B39]+的最稳定结构(具有C3对称性)为初始结构进行几何优化。在PBE0/6-311+G*/RECP理论水平下优化了所有[An@B39]n+的结构,所有体系均计算了不同自旋态。对于[U@B39]3+,考虑了单重态、三重态、五重态和七重态;而对于[Cm@B39]2+,则考虑了二重态、四重态、六重态和八重态。因此,每个体系计算得到了四个异构体。
图1列出了[U@B39]3+和[Cm@B39]2+的所有异构体的结构和相对能量。与预期结果一致,[An@B39]n+的能量最低异构体均为内嵌硼球烯结构。[U@B39]3+的最稳定结构为三重态,且具有C3对称性;而[Cm@B39]2+为八重态,且不具有对称性(C1)。[U@B39]3+与[Cm@B39]2+的对称性不同,这可能是由于U和Cm不同的原子半径导致的。对于每个体系的最稳定结构,硼笼在顶部和底部均为六边形孔洞,而在腰部具有四个七边形孔洞。与B-39类似,整个硼笼由46个硼三角形组成。

图1 优化得到的[An@B39]n+异构体的结构图和相对能量Fig.1 Optimized geometrical structures of[An@B39]n+and relative energy
2.2 [An@B39]n+的成键性质分析
[An@B39]n+体系中An-B键距、中心金属原子上的原子电荷(QAn)以及单电子最高占据轨道(SOMO)/HOMO-LUMO能隙列入表1。由表1可知,在DFT-PBE0理论水平下优化得到的[U@B39]3+最稳定结构中,U-B键的距离为2.86~3.43Å(1Å=0.1 nm),并且平均键距为3.15Å。对于[Cm@B39]2+,最短和最长的Cm-B键距分别为2.80Å和3.68Å,平均键距(3.24Å)较U-B键稍长。对于所有体系,An-B键距均大于U(1.70Å)、Cm(1.66Å)和B(0.85Å)的共价单键半径之和[48]。这些较长的键距表明,[An@B39]n+可能存在多中心离域键[49]。
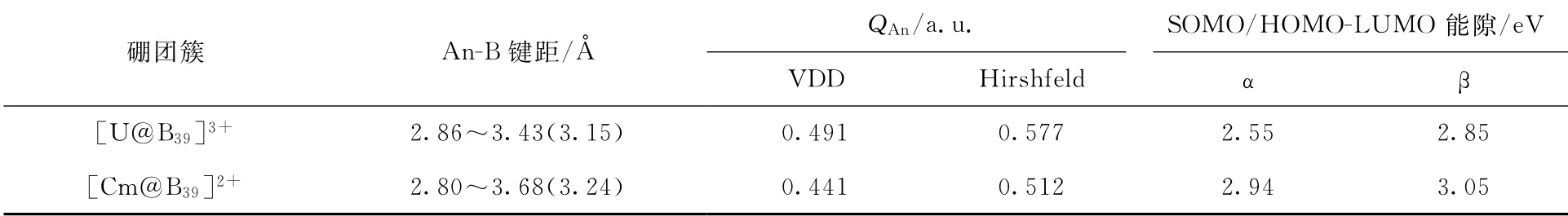
表1 [An@B39]n+体系中An-B键距、中心金属原子上的原子电荷(Q An)以及SOMO/HOMO-LUMO能隙Table 1 An-B bond distance of[An@B39]n+,atomic charge(Q An)on central metal atom and SOMO/HOMO-LUMO gap
为了评估[An@B39]n+的电荷重排,在PBE0/6-311+G*/RECP理论水平下进行了VDD[46]和Hirshfeld[50]电荷分析(表1)。Hirshfeld电荷受基组的影响较小,具有很强的物理意义并且计算速度较快[50],而VDD电荷主要用于描述化学键导致的电荷的流动[51]。[U@B39]3+的中心金属原子上的VDD和Hirshfeld电荷分别为0.491 a.u.和0.577 a.u.,而[Cm@B39]2+的相应值为0.441、0.512 a.u.。这表明存在从U或Cm到B-39笼的电荷转移,这与B具有较高的电负性一致。如表1所示,在PBE0/6-311+G*/RECP理论水平下,计算了SOMO/HOMO与LUMO之间的能隙。[An@B39]n+的SOMO/HOMO-LUMO能隙相对较大,说明这些体系均比较稳定。
为了深入地了解内嵌的锕系金属原子与硼笼之间的相互作用,在PBE0/6-311+G*/RECP理论水平下对[An@B39]n+进行了Ad NDP分析(图2)。Ad NDP分析在硼团簇体系中得到了广泛的应 用[25,27,47,52-55],主 要 用 于 揭 示 体 系 中 的 多中心离域键,并可以获得相应的多中心轨道[42]。[U@B39]3+配合物具有59对自旋成对的价电子和两个单电子,根据Ad NDP分析(图2),体系中共有59个离域的σ键和π键。其中包括39个3c-2eσ键位于39个B3三角形上,4个6c-2eσ键,以及1个7c-2eσ键。此外,在体系中,同时存在3个4c-2eπ键、9个5c-2eπ键、2个7c-2eπ键和1个39c-2eπ键,这些离域π键显示了锕系金属与硼原子之间的相互作用。因此,[U@B39]3+的120个价电子中有118个价电子参与离域的σ键或π键。同样,分析发现[Cm@B39]2+125个价电子中118个价电子参与了离域的σ键或π键。如上所述,所有这些锕系硼球烯体系均存在σ和π离域键,并且锕系金属内嵌硼球烯中金属与硼笼之间存在共价相互作用。

图2 [U@B39]3+的Ad NDP分析Fig.2 Ad NDP analysis of[U@B39]3+
在PBE0/6-311+G*/RECP理论水平下,采用QTAIM分析进一步探讨[An@B39]n+的成键性质。QTAIM基于波函数分析,具有明确的数学意义,并且对基组敏感性低[56]。QTAIM分析中,在两个原子之间的键临界点(BCP)处,成键性质可以通过电子密度(ρ)和拉普拉斯电子密度(Δ2ρ)以及能量密度(H)来反映。通常,在BCP处,ρ>0.20 a.u.,且Δ2ρ为负值时,化学键为共价键;若ρ<0.10 a.u.,Δ2ρ为正值,则表示闭壳层非共价相互作用(离子键、范德华相互作用和氢键等)。H为负值时,表示共价相互作用,其大小反映了键的共价程度。在BCP处,H<0和Δ2ρ>0对应部分共价相互作用[57-58]。[An@B39]n+的QTAIM分析结果列入表2。由表2可知:对于[U@B39]3+,计算得到6个BCPs,而[Cm@B39]2+仅有3个BCPs,表明[An@B39]n+中锕系原子和硼原子之间存在相互作用,且U-B相互作用更强。[U@B39]3+和[Cm@B39]2+的ρ值均非常小,表明其共价相互作用较弱。尽管[An@B39]n+的Δ2ρ均为正值,但是其H值均为负。因此,[An@B39]n+中的An-B键为部分共价相互作用。另外,[U@B39]3+的H值比[Cm@B39]2+的更负,也表明U-B键较强。
电子局域函数(ELF)是一种广泛用于测量电子局部化程度的函数[59]。电子在特定区域中的定域程度越高,内部电子越难以使其区域离域,并且相应地,外部电子越难以进入[44]。ELF值越高,电子定域的程度越高,表明所涉及的电子具有共价键[60]。如表2所示,U-B键显示出比Cm-B键更大的ELF值,表明铀内嵌硼球烯的共价键成分更高。

表2 [An@B39]n+的QTAIM分析Table 2 QTAIM analysis of[An@B39]n+
前线轨道(FMO)理论指出,HOMO的能级越高,电子成为亲电位点的可能性就越大[44]。在PBE0/6-311+G*/RECP理论水平下,对[An@B39]n+进行了MO分析,结果示于图3。根据MO分析,对于[U@B39]3+,MOs(HOMO-1(a)、HOMO-2(e)、HOMO-3(e)和HOMO-4(a))为U-B成键轨道,主要是由U 5f和B 2p轨道贡献而成。而轨道SOMO(e)和SOMO-1(e)基本是由B 2p轨道的相互作用引起的。另外,[U@B39]3+的MO的组成要大于[Cm@B39]2+的。例如,[U@B39]3+的HOMO-1(a)、HOMO-2(e)和HOMO-3(e)的U-B键U 5f轨道贡献成分分别为23%、27%和27%,比[Cm@B39]2+的Cm-B成键轨道SOMO-3(a)、SOMO-4(a)和SOMO-5(a)的5f轨道成分(5%、15%和15%)大得多。此外,如图3所示,[U@B39]3+的轨道能级总体比[Cm@B39]2+的轨道能级低,这表明U-B键的相互作用比Cm-B键更强。当然,另一方面,因为[U@B39]3+团簇相较[Cm@B39]2+团簇多了一个正电荷,从而其也有利于具有能量更低的前线分子轨道。因此,在这些锕系金属内嵌硼球烯中,锕系元素和硼原子之间存在直接相互作用,并且U-B键比Cm-B键具有更强的共价相互作用,这与前述分析结果一致。
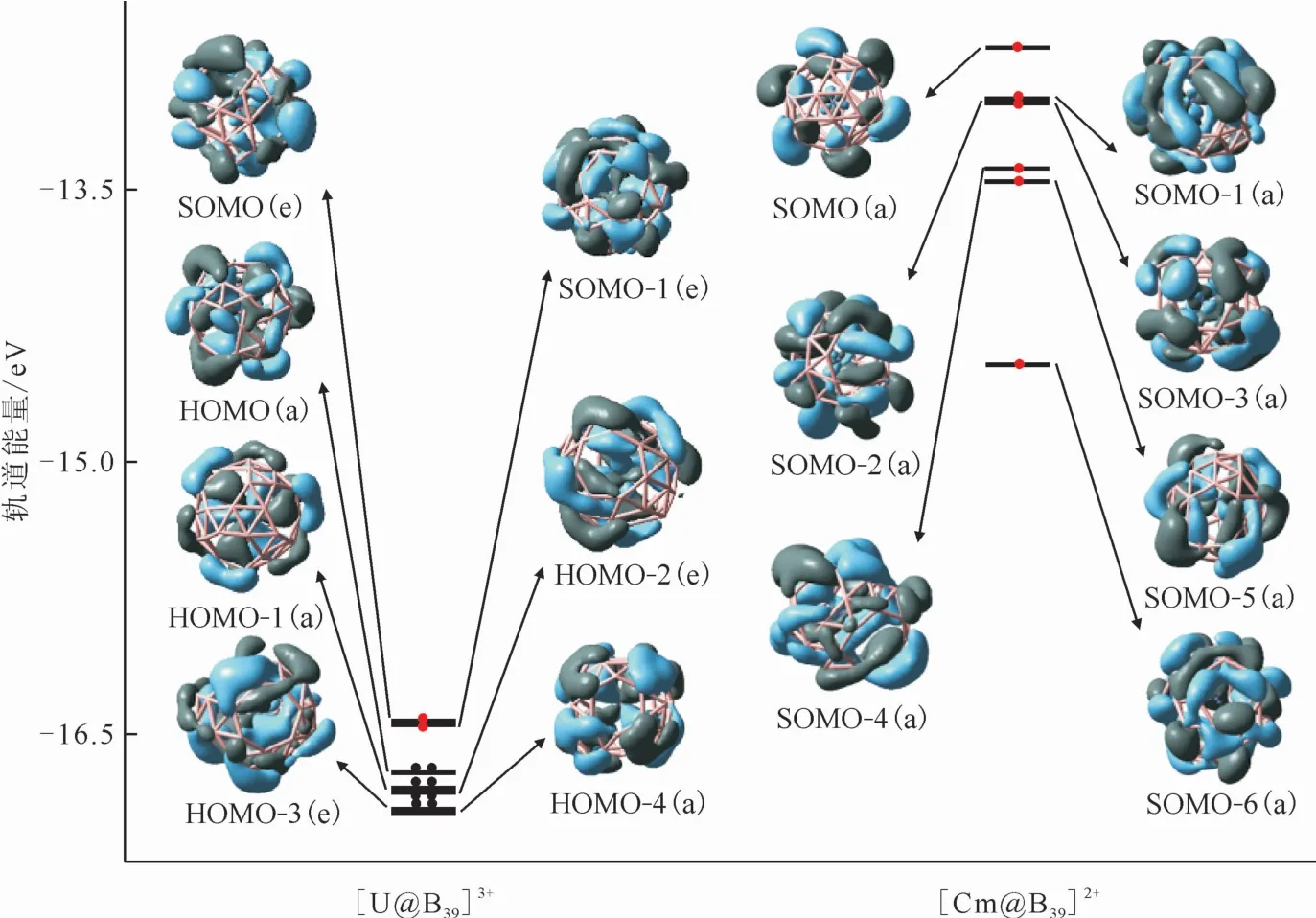
图3 [An@B39]n+的前线分子轨道图Fig.3 Frontier molecular orbitals of[An@B39]n+
根据文献[61]报道,结合红外(IR)光谱和第一性原理计算是表征团簇的有效方法。在PBE0/6-311+G*/RECP理论水平下对[An@B39]n+进行了振动频率分析并模拟了体系的红外光谱(图4)。为了进行比较,在相同的理论水平下计算了C3B3-9的红外光谱。如图4所示,[U@B39]3+在625、1 043 cm-1和1 248 cm-1具有3个主 要 的IR峰,与C3B3-9(537、1 035 cm-1和1 234 cm-1)相似。与[U@B39]3+类似,[Cm@B39]2+也显示了相似的红外特征峰,这表明所有[An@B39]n+结构中硼笼均基本保持了B-39的光谱特征。通过对比模拟的红外光谱,在[An@B39]n+中也发现了144 cm-1以下的IR峰,这对应于锕系金属原子和硼原子之间的红外振动频率,证明An-B之间的共价相互作用。对于模拟的[An@B39]n+拉曼光谱(图5),[U@B39]3+和[Cm@B39]2+的特征峰均与C3B-39非常接近,而且均能观测到An-B键的振动峰。

图4 理论模拟的[An@B39]n+红外光谱Fig.4 Simulated infrared spectra of[An@B39]n+
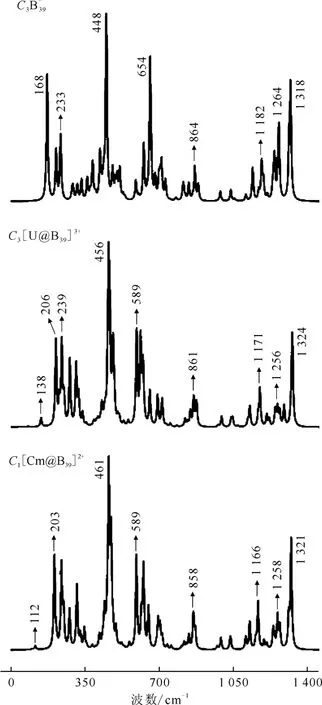
图5 理论模拟的[An@B39]n+拉曼光谱Fig.5 Simulated Raman spectra of[An@B39]n+
在PBE0/6-311+G*/RECP理论水平下,计算了[An@B39]n+的形成能以评估这些锕系内嵌硼球烯的相互作用及热力学稳定性,结果列入表3。根据计算结果,[U@B39]3+和[Cm@B39]2+的形成能均为负值,且绝对值相对较高,其值分别为-1 095.0 kcal/mol和-773.3 kcal/mol(1 kcal=4.187 kJ),比文献[47]报道的[Ca@B39]+的形成能(-299.3 kcal/mol)更负,表明这些锕系内嵌硼球烯均具有较高的热力学稳定性。此外,[U@B39]3+具有更负的形成能,与此体系较强的U-B共价键一致。由此,推断锕系金属原子与硼笼之间的共价相互作用可能对[An@B39]n+的形成至关重要。

表3 [An@B39]n+的形成能Table 3 Formation energy of[An@B39]n+
3 结 论
在DFT-PBE0理论水平下系统研究了一系列锕系金属内嵌硼球烯[An@B39]n+(An=U,n=3;An=Cm,n=2)。结果表明,U和Cm掺杂的硼球烯([U@B39]3+和[Cm@B39]2+)均能形成稳定的金属内嵌硼球烯。具有C3对称性的结构是[U@B39]3+的最稳定结构,而[Cm@B39]2+则为C1结构。根据Ad NDP分析,[U@B39]3+和[Cm@B39]2+存在离域的σ键和π键,与Cm-B键相比,U-B键的共价性成分相对较高。这些结果与QTAIM和MO分析结果一致。不出所料,[An@B39]n+的红外光谱和拉曼光谱均能观测到An-B键的振动峰。本工作预测的这些锕系金属内嵌硼球烯不仅丰富了硼团簇的种类,而且为设计新型金属内嵌硼球烯提供了理论基础。
猜你喜欢
药学进展(2022年1期)2022-03-06
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
潍坊学院学报(2021年2期)2021-07-22
空间科学学报(2020年6期)2020-07-21
空间科学学报(2020年6期)2020-01-08
环球时报(2019-12-05)2019-12-05
食品科学(2019年14期)2019-07-26
物理化学学报(2015年7期)2015-12-30
