
钕铁硼废料中氟含量的测定
——离子选择电极法
2022-11-05 07:12姚南红龚雪颍温斌
稀土信息 2022年9期
姚南红 龚雪颍 温斌/文
钕铁硼废料在收集、存储和贸易过程中,会被混入稀土熔盐渣,导致钕铁硼废料也有氟的存在。在钕铁硼废料加工回收过程中,氟与稀土离子形成氟化物沉淀随废渣排放而导致收率降低,且氟离子进入萃取工艺而影响稀土分离。再者,氟若不加以处理就排放的话,既浪费了氟资源,又污染了环境,氟污染具有强穿透性和不可逆转性。准确、快速地分析钕铁硼废料中氟含量并加以回收利用是氟污染控制、治理和资源化的一个重要步骤。
氟量的检测方法主要有离子选择电极法、EDTA滴定法及色谱法。本文采用了离子选择电极法分离稀土测定钕铁硼废料中氟含量,方法简单快捷、准确可靠。
原理:灼烧后钕铁硼废料经硝酸、高氯酸分解,保持在130 ℃~140 ℃通入水蒸气进行蒸馏,使氟与其他共存元素分离。馏出液调整pH值为5.5~6.0,加入总离子强度缓冲溶液,以氟离子选择性电极为指示电极,饱和甘汞电极为参比电极。测量两电极间的平衡电位值,计算氟含量。
一、实验部分
1.仪器与试剂
(1)仪器:离子计
(2)总离子强度缓冲溶液TISAB(pH≈5.5~6.0):称取15.0 g 乙酸钠,60.0 g 柠檬酸三钠,17.0 g 氯化钠,8.0 g 乙二胺四乙酸二钠盐于1000 mL 烧杯中。加入500 mL 水,搅拌至溶解清亮,加入3 mL 乙酸,移入1000 mL 容量瓶中,用水稀释至刻度,混匀。立即保存于塑料瓶中。
(3)硝酸、高氯酸为分析纯,试验用水为Ⅱ级水。
2.试验方法
(1)样品制备及检测
称取炉渣料、块片料、干燥粉料、油泥料、潮湿粉料等钕铁硼废料样品 约50 g,精确至0.01 g,置于100 mL 已在950 ℃烧至恒重的瓷蒸发皿中,在300 ℃~400 ℃电热板上加热灼烧0.5~1 h 至干燥,稍冷,置于干燥器中冷却至室温,称重,立即研磨均匀。
称取上述灼烧后钕铁硼废料试样1.0 g 精确至0.0001 g。按图1 所示安装蒸馏分离装置,将试料置于500 mL 蒸馏瓶中,加入10 mL 水润湿试料,加入5 mL(1+1)硝酸硝化,待1~2 min 硝化反应完全后,加20 mL 高氯酸。通入冷凝水,以盛有2 mL 氢氧化钠溶液(100 g/L)的250 mL 烧杯吸收蒸馏液。加热冷凝水,待温度升至120 ℃~ 130 ℃时通入水蒸气,控制蒸馏瓶中液相温度为130 ℃~140 ℃,蒸馏速度控制为5~6 mL/min。待馏出液体积约为200 mL时,停止蒸馏,将馏出液转移至250 mL 容量瓶,用水稀释至刻度,混匀。移取10 mL 馏出液于50 mL容量瓶中,加入2 mL 氢氧化钠溶液(100 g/L),加1 滴溴甲酚绿指示剂,用盐酸(1+10)调至溶液呈黄色,再用氢氧化钠溶液(10 g/L)调至溶液显蓝色,加10 mL 总离子强度调节缓冲溶液,以水稀释至刻度,混匀。
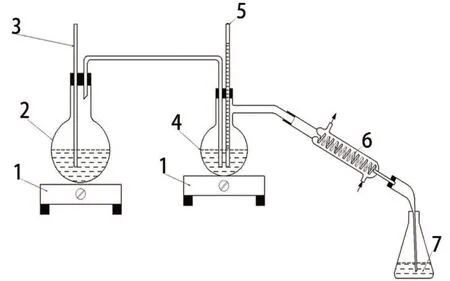
图1 蒸馏分离装置示意图
将上述溶液全部转移至50 mL 干燥的塑料烧杯中,加入磁力搅拌转子,放置电磁搅拌器上,启动搅拌器。插入已清洗干净且干燥的氟离子选择电极和饱和甘汞电极,待其读数稳定,电极电位变化每分钟不大于0.2 mV 时,读取电位值。在工作曲线上查出氟含量。随同试样做空白试验。
(2)氟离子标准工作曲线的绘制
分别移取0.50 mL、1.00 mL、2.50 mL、5.00 mL、7.50 mL、10.00 mL、20.00 mL 氟离子标准溶液(1 mg/100 mL)于50 mL 容量瓶中。按氟浓度由低到高的次序与试料同时进行测定。
以氟量为横坐标,以相对应的平衡电位值为纵坐标,绘制半对数工作曲线。
(3)结果的计算

式中:
m1——从标准曲线上查得分析试液氟量,单位为微克(μg);
m0——随同空白中氟量,单位为微克(μg);
V0——试液总体积,单位为毫升(mL);
m2——试料的质量,单位为克(g);
V1——分取试液体积,单位为毫升(mL);
m3——瓷蒸发皿及试料灼烧后的质量,单位为克(g);
m4——瓷蒸发皿质量,单位为克(g);
m5——试料灼烧前的质量,单位为克(g)。
两次平行测定结果的绝对差值不大于表1 中相应重复性限时,取其平均值为测定结果。

表1 重复性限
二、结果与讨论
1.样品处理方式的选择
分别称取1#、2#、3#、4#各4 份,其中两份按本试验方法进行蒸馏、加标蒸馏进行测定。另两份分别进行碳酸钠-过氧化钠熔融、熔融加标测定。两种样品处理结果及加标回收率见表2。

表2 不同样品处理方式的分析结果
上表数据表明,熔融法测定钕铁硼废料中氟的方法因样品中存在较多杂质干扰元素(如硼、钙、镁、钴等)无法完全掩蔽而不能满足检测的要求。蒸馏法因使被测元素氟完全与样品基体分离而较好的排除了干扰元素的影响。故实验选择蒸馏法进行样品处理。
2.总离子强度缓冲溶液pH 值
使用氟标准溶液在不同pH 值的TISAB 中进行测定并绘制标准工作曲线,实验结果见表3。

表3 不同样品处理方式的分析结果
通过对比曲线回归方程和相关系数得出最佳的pH 值为6。pH 值对离子选择电极测定氟离子浓度有较大影响,pH < 5.5 溶液中会发生下述弱酸配位反应:2F-+H+=HF+F-=HF2-,使溶液中的F-减少,会影响电极的灵敏度,使分析结果偏低。当pH >7时,OH-对电极的响应,将严重影响测定结果,使分析结果偏高。分析测定时pH值需控制在6左右(每次配置新的TISBAB 后都需要重新绘制标准工作曲线)。
3.高氯酸、氢氧化钠用量的选择
准确分取5000 ug 离子标准贮存溶液于盛有不同体积高氯酸的500 mL 蒸馏瓶中,分别加入不同体积的氢氧化钠,按照实验方法对其蒸馏、测定并计算回收率,实验结果见表4。

表4 不同高氯酸、氢氧化钠用量的结果
实验表明蒸馏体系高氯酸量为15 mL~ 25 mL时氟回收率趋于稳定。所以实验选择高氯酸的加入量为20 mL,吸收液体积对结果没有明显影响,本实验选择2 mL。
4.蒸馏时间、溶液温度对测氟的影响
分取5000 ug 氟离子标准贮存溶液于盛有20 mL高氯酸的蒸馏瓶中。按照实验方法对其测定并计算回收率,实验结果见表5。

表5 不同蒸馏时间、温度对结果的影响
实验表明蒸馏过程中,溶液温度控制在130℃~160℃,蒸馏时间控制在30 min~40 min时,蒸馏体系中的氟蒸馏完全。但温度过高会产生大量高氯酸浓烟,所以实验选择温度控制在130℃~140℃。
5.总离子强度缓冲溶液加入量的选择
氟离子测定时,被测溶液的pH 影响测定准确度的因素,移取1#和4#各4 份于50 mL 的容量瓶中,按表6 加入不同体积的缓冲溶液,按方法步骤对缓冲溶液加入量实验。

表6 总离子强度缓冲溶液加入量的选择
从表6 看出,当缓冲溶液加入体积10~15 mL时,测定结果较稳定,本实验选择总离子强度缓冲溶液加入量10 mL。
6.标准曲线的绘制

图2 氟标准半对数曲线
按氟离子标准曲线方法绘制标准曲线,见图2。经过多次试验,曲线相关性系数r均大于0.9998,能满足分析测定的需要。
7.空白和检测下限试验
在本试验的条件下,测定上限按照标准系列略低于标准曲线最高点的原则,试验确定检测上限为1.0%。
试验对随行空白进行11 次平行测定,标准偏差(δ)小于0.0010%,以10 倍空白值的标准偏差作为检测下限,考虑到实际样品的基体的差异性,确定试验方法的测定范围为0.010%~1.0%是合适的。
8.精密度实验
分别称取1#、2#、3#、4#、6#,按方法步骤对统一样进行11 次重复测定,统计平均值和相对标准偏差考察方法精密度,实验数据汇总如表7。
9.加标回收实验
按试验步骤要求分别称取1#、2#加入硝酸、高氯酸后,分别加入氟离子标准溶液(100μg/mL)2.50 mL、8.00 mL,按试验步骤要求分别称取3#、4#加入硝酸、高氯酸后,分别加入氟离子标准溶液(1000μg/mL)2.0 mL、2.5 mL,按实验步骤进行回收率实验;结果见表9。
由表7 和表8 的结果可见,标加回收率实验数据为96.70~104.80%,RSD < 7 %,表明本方法的准确度和精密度令人满意。


表7 统一样数据汇总
三、结论
采用本方法能够解决钕铁硼废料中氟元素与其他共存元素分离困难的问题,不仅为钕铁硼废料的氟含量的测定提出了新的方法,而且为钕铁硼废料稀土回收以及氟污染的控制、治理和资源化利用提供了一定的思路借鉴。本方法确定了钕铁硼废料中氟含量测定的最佳分析条件。方法准确可靠,操作简单,精密度和准确度均能满足分析的要求。

表8 回收率实验数据汇总
猜你喜欢
稀土信息(2021年3期)2021-04-03
中欧商业评论(2020年12期)2021-01-09
化学与粘合(2020年6期)2020-03-08
探索科学(学术版)(2019年5期)2019-07-13
山西冶金(2018年1期)2018-03-28
科技视界(2017年25期)2017-12-11
河北地质(2016年2期)2016-03-20
无机化学学报(2014年6期)2014-02-28
河南科技(2014年15期)2014-02-27
中国无机分析化学(2012年4期)2012-12-01
