
Phelan- McDermid综合征患儿1例并文献复习
2022-11-03 04:03罗举语林艳张占会林舜娜李冰肖
河南医学研究 2022年20期
罗举语,林艳,张占会,林舜娜 ,李冰肖
(1.暨南大学附属第一医院 a.神经内科;b.儿科, 广东 广州 510630;2.天河区妇幼保健院 儿科,广东 广州 510610)
Phelan-McDermid综合征(PMS)也称22q13.3缺失综合征,是22号染色体长臂末端缺失导致的罕见遗传性疾病,显性遗传,发病率无明显性别差异[1]。PMS是神经发育障碍性疾病,其临床特征为新生儿肌张力低下、全面性发育迟缓、中重度的智力障碍严重的语言延迟或无语言、孤独症谱系障碍和轻微的面部畸形,其他特征包括肥大的手、发育不良的趾甲、淋巴水肿和疼痛感降低,部分患者还会出现斜视、听力损伤、肾功能障碍、癫痫、蛛网膜囊肿等[2]。
PMS主要是由缺失区内SHANK3功能丧失(或单倍体功能不全)或点突变引起[3-4]。引起PMS 的22q13.3缺失片段大小长短不一,短缺失可小于100 kb,最长可延伸至大于9 Mb[5]。染色体片段缺失包括简单缺失、不平衡易位、环状染色体形成或涉及其他22号染色体结构重排等[6]。几乎所有的缺失片段都涉及SHANK3基因[7]。SHANK3也被称为ProSAP2,在大脑中的许多区域均有表达。SHANK3基因编码谷氨酸能突触后致密区的支架蛋白,该支架蛋白通过调节树突的形成在突触功能中发挥关键作用[8]。
本例患儿染色体全基因组高通量测序结果显示,染色体chr22:4 65194 80-51304 566上存在4.79 Mb的新发缺失突变,缺失区间包含了完整的SHANK3基因;临床表现全面发育迟缓,语言发育落后,肌张力低下,耳大,大而肥的手,发育不良的趾甲,因此诊断为PMS,现将其诊疗过程和诊断依据报告如下。
1 资料与方法
1.1 研究对象患儿,女,2岁,因“全面性发育落后”从8月龄开始在暨南大学附属第一医院行康复训练1年余,仍存在肌张力低下,语言及运动发育落后,严重精神发育迟缓及孤独症样表现。高度疑似遗传性疾病。患儿父母对本研究内容知情同意。本研究通过广州市天河区妇幼保健计划生育服务中心医学伦理委员会审核批准。
1.2 全基因组高通量测序抽取患儿及其父母外周静脉血各2 mL,经乙二胺四乙酸抗凝,送往北京智因东方转化医学研究中心进行家系3人全基因组测序。
1.3 文献检索以“Phelan-McDermid综合征” “22q13.3缺失综合征”“SHANK3基因”为关键词,在中国知网、万方数据库检索自建库截至2021年12月于国内发表的基因确诊的病例报道;以“Phelan-McDermid syndrome”“22q13 deletion syndrome”“SHANK3gene”为关键词,在PubMed检索自建库截至2021年12月于国外发表的文献。纳入标准:(1)临床资料完整;(2)与SHANK3基因相关的PMS且经基因诊断确诊;(3)中国人。排除产前诊断为PMS的患者。系统总结所有检索到的病例的临床特点及其发病率。
2 结果
2.1 临床发现患儿系第2胎第2产,足月剖宫产,出生体质量3.75 kg,出生时无窒息史,父母非近亲结婚,否认遗传病家族史。8月龄时因“肌张力低下,不会坐”就诊,诊断为全面性发育迟缓。从9月龄开始在暨南大学附属第一医院接受神经肌肉电刺激及运动康复训练。1岁半时能独坐及腹爬,2岁时会手膝爬,会扶站。双手抓握力量弱,抓到玩具即刻丢掉,不会讲话,理解差,目光对视差,无应名反应。有刻板行为,表现为拿到食物咬一口马上丢掉,再捡起来咬一口再丢掉。2岁体格检查:身高89.3 cm(第97百分位数),体质量12.5 kg(第99百分位数),头围49.7 cm(第97百分位数),耳大,手足肥大,骶骨酒窝,见图1。心、肺、腹部查体无异常,四肢肌张力降低,痛感下降,肌力4级,腱反射正常,病理反射未引出。
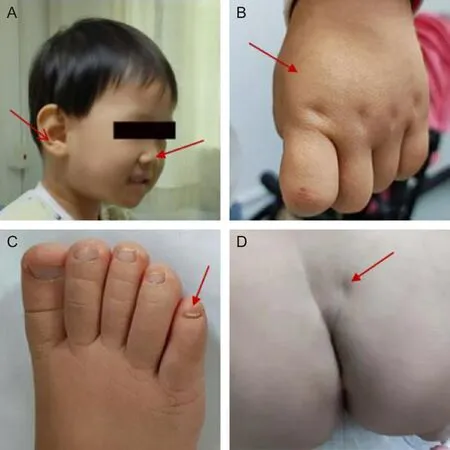
A.耳大、宽鼻梁;B.大而肥的手;C.发育不良的趾甲;D.骶骨酒窝
2.2 辅助检查2岁龄时0~6岁小儿神经心理发育评估结果:大运动9.5个月,精细动作9个月,适应能力8.5个月,语言8个月,社交行为9个月,发育商36分。头颅MRI提示双侧半卵圆中心白质发育较少。脑电图、听力脑干反应结果均正常。胸部、髋关节、足部X检查均未见异常。血常规、生化、电解质、肝肾功能、25-羟基总维生素D、甲状腺功能均正常。血尿遗传代谢筛查未见异常。
2.3 全基因组高通量测序结果高通量测序检测结果显示患者存在拷贝数变异常,为新发杂合缺失;缺失在染色体上的位置为chr22:46 519 480-51 304 566,大小为4.79 Mb,见图2。未发现患儿父母有该缺失变异。患儿缺失片段内包含TYMP、CELSR1、SBF1、ARSA、TUBGCP6、MLC1、TRMU、SCO2、ALG12、SHANK3、CHKB基因。
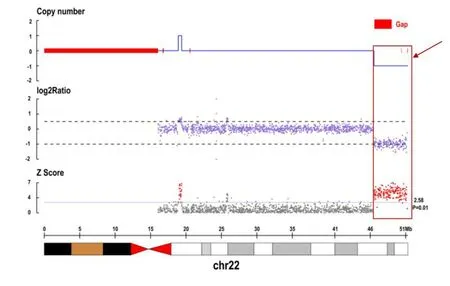
图2 先证者(女,患儿)在chr22染色体拷贝数分布图
2.4 文献复习结果以“Phelan-McDermid综合征”“22q13.3缺失综合征”和“SHANK3基因”为关键词,在中国知网、万方数据库检索到6例中国患者;以“Phelan-McDermid syndrome”“22q13 deletion syndrome”“SHANK3 gene”为检索词,检索到34例中国患者,共涉及国内外文献9篇[9-17],加上本例患儿共41例PMS患者。PMS患儿发育、行为特征见表1;PMS患儿畸形特征见表2;PMS患儿基因变异特点见表3。
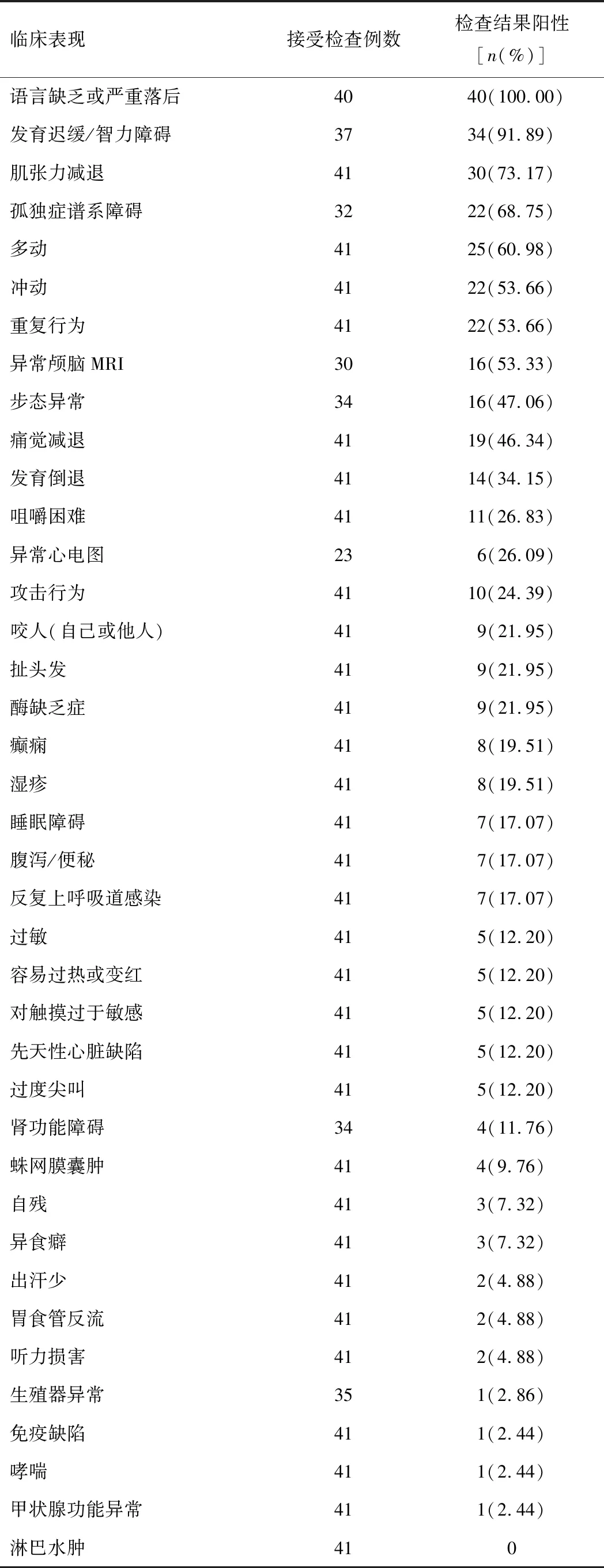
表1 PMS患儿发育、行为特征

表2 PMS患者畸形特征

表3 患儿基因变异特点
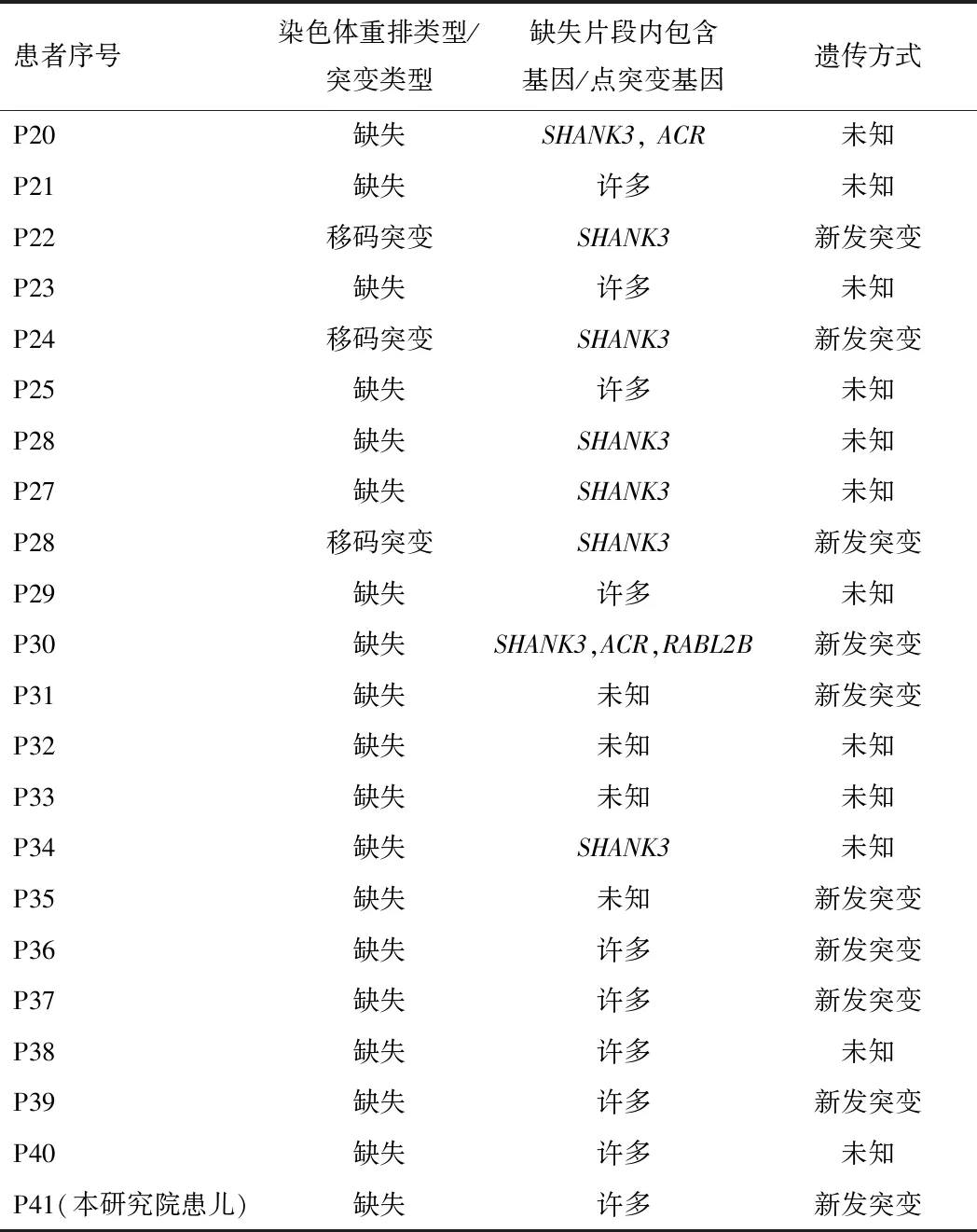
表3(续)
3 讨论
1985年,Watt等[18]首次报道了1名14岁PMS男患儿,其临床表现为语言缺失、轻微畸形特征,严重智力残疾,被认为是母源22号染色体臂间倒位使得减数分裂重组,导致22q12至22q末端缺失。PMS又被称为22q13.3缺失综合征,因为大多数PMS病例主要是由22q13.3缺失引起,但也有许多基因缺失,其中,SHANK3基因与PMS神经学特征关系最为密切。SHANK3单倍剂量不足是导致PMS的主要原因[3,9-10,19],即22q13.3区域的缺失导致PMS患者的每个细胞SHANK只有1个拷贝数,而不是正常的2个拷贝数[20]。PMS临床表现广泛,神经系统表现包括发育迟缓、智力障碍、言语缺失或发育落后、癫痫、皮肤过热或变红、出汗减少、对触摸过度敏感、痛觉减退、蛛网膜囊肿、低肌张力减退、步态异常、孤独症谱系障碍、睡眠障碍、发育倒退等;畸形特征主要有小头症(头小畸形)、巨头症、稀疏眉毛、长睫毛、眼距过宽、宽鼻梁、蒜头鼻、耳畸形(耳大)等;其他表现为胃食管反流、腹泻或便秘、生殖器异常、湿疹、免疫缺陷、反复上呼吸道感染、听力损失、先天性心脏缺陷、肾功能障碍、哮喘等[21]。本研究患儿染色体chr22:46 519 480-51 304 566上存在4.79 Mb的新发缺失突变,缺失区间包含完整的SHANK3基因及其他11个致病基因,其中SHANK3关联PMS和精神分裂症15型,SCO2基因关联近视6型,CELSR1基因关联淋巴管畸形9型,这3个关联疾病均是常染色体显性遗传。本例患儿具有典型PMS的临床表现,如发育迟缓、智力障碍、语言缺乏、肌张力降低、轻微面部畸形特征(耳大,长睫毛、宽鼻梁)、痛觉减退、孤独症样行为、大而肥的手、发育异常的趾甲、骶骨酒窝、身材高大和痛觉减退,但患儿无妄想、幻觉,近视,淋巴水肿的临床表现,因此排除精神分裂症15型、近视6型和淋巴管畸形9型。综上所述,结合临床表现及分子遗传学检测结果,该患儿可确诊为PMS。
基因型与表型研究表明,染色体片段缺失的大小与临床表现多样性和/或严重程度呈正相关[5,22-25],如与新生儿低肌张力[25]、发育迟缓[26]、畸形特征[25]、言语能力[5]、孤独症相关的社交障碍[2]以及其他临床表现[2]。此外,末端缺失较小的患者可能比缺失较大的患者发育更好[27]。有研究还表明,缺失所包含的额外基因也有助于表型表达[27]。但是,不同文献之间基因型与表型研究结果仍未达成共识[4,22,28-29]。近年来,随着大规模的测序技术的发展及应用,SHANK3基因点突变引起的PMS患者不断被检测出来,有研究发现[4],由点突变引起的SHANK3单倍剂量不足以导致一系列与PMS相关的症状,且与片段缺失的患儿相比,点突变患儿言语障碍和运动障碍更轻,提示其他基因缺失也参与了PMS的病因学。
目前,PMS的临床诊断标准尚未完全建立,主要基于典型临床表现和分子遗传学检测[30]。分子遗传学检测主要包括染色体微阵列、单基因检测、全基因组高通量测序等。本例患儿出生时体质量、身长、头围均正常,生长速度高于平均水平。2岁时身长、体质量、头围均超过同性别同年龄儿童95百分位数。婴儿期存在肌张力及肌力低下,运动发育落后,语言理解及表达障碍,刻板行为及轻微面部畸形。由于出生时无窒息史,无颅内感染史,头颅MRI仅表现为脑白质髓鞘发育延迟,脑电图及遗传代谢筛查均无异常。经过 1 a多康复训练,孩子运动发展非常缓慢,而认知水平则停止在8月龄水平。而患者轻微的面部畸形极易被临床医生忽略。这说明PMS的临床表现多样且缺乏特异性,容易误诊为脑瘫、精神发育迟缓、孤独症谱系障碍。因此,及时的分子遗传学检查对PMS确诊非常重要。
本例患儿经过康复训练后在2岁时复诊时会扶站及移动数步,肌张力降低,步态异常,会发“mama”音,肌力4级,但也有文献报道,未经任何康复训练的多数患儿在3岁以后也能独立行走[9,31]。因此,PMS患者是否必须经过康复训练才能独立行走是一个值得商榷的问题。已有文献报道,淋巴水肿只存在于大于10岁且缺失大于4.3 Mb的患者中,提示淋巴水肿可能与年龄相关[5]。本研究患儿缺失片段为4.79 Mb,目前尚无淋巴水肿的表现,但医生一直警惕发生淋巴水肿的可能。缺失大于1.3 Mb的患者常伴有肾功能障碍[24],UPK3A,ZBED4,CELSR1和FBLN1被认为是导致肾脏异常的候选基因[29,31]。本研究该患儿虽然缺失片段也涉及CELSR1基因,但目前并未出现肾功能障碍。
综上所述,本研究通过临床表现和分子遗传学的检测确诊了1例PMS患儿。虽然基因型与表型关联研究结果不一致,但缺失的大小与临床表现多样性和/或严重程度呈正相关,同时缺失片段中包含的其他基因缺失也参与PMS的病因学。
猜你喜欢
中华实用诊断与治疗杂志(2022年1期)2022-08-31
现代仪器与医疗(2022年1期)2022-04-19
科学之谜(2019年3期)2019-03-28
科学之谜(2018年8期)2018-09-29
家庭百事通·健康一点通(2017年8期)2017-08-18
中外医学研究(2016年35期)2017-02-28
中学生理科应试(2016年4期)2016-11-19
恋爱婚姻家庭·养生版(2016年9期)2016-09-07
中国实用医药(2016年9期)2016-05-17
中学生英语高效课堂探究(2011年4期)2011-07-07
