
可见光/CuFeO2/H2 O2 体系降解氧氟沙星
2022-11-01 11:21张立东霍思月高孟春
兰州交通大学学报 2022年5期
张立东,霍思月,李 杰,高孟春
(1.兰州交通大学环境与市政工程学院,兰州 730070;2.吉林化工学院资源与环境工程学院,吉林吉林 132022;3.中国海洋大学海洋环境与生态教育部重点实验室,山东青岛 266100)
氧氟沙星(OFX)属于喹诺酮类抗生素,由于其具有较宽的抑菌谱、较强的活性和较少的不良反应被广泛用于家畜疾病控制[1-2].近年来,OFX类抗生素的大量生产和过度滥用对生态环境产生了深远的影响,并对人类健康构成一定威胁,因此,寻找先进的处理技术以降低OFX类抗生素的毒性,减轻其对生态环境和人类健康的威胁变得尤为重要[3].目前,抗生素类处理技术包括物理法[4]、生物法[5]和高级氧化技术(AOPs)[6].物理法主要包括膜分离和吸附,其成本较高且存在二次污染问题[4].生物法有好氧生物处理和厌氧生物处理等,但抗生素普遍存在生物毒性,易使微生物和细菌产生耐药性[5].鉴于此,芬顿高级氧化技术受到人们广泛关注,其能在电、光、催化剂等反应条件下产生高效活性氧物种(ROS),将大部分难降解有机物矿化成无机化合物或小分子物质进而达到高效去除污染物的目的[7].
传统均相芬顿法具有降解效率高、工艺简单和绿色环保等特点,主要原理是利用液相中的Fe2+和H2O2在酸性条件下反应产生高活性·OH氧化难降解有机物使污染物得以高效去除[8].然而此方法对pH值要求苛刻,还会产生大量的污泥[9].与之相比,异相类芬顿法使用固相铁基催化剂代替溶解态Fe2+,具有pH适用范围宽、二次污染少以及催化剂可重复利用的优势[10].然而其反应过程中单一铁矿物表面的Fe3+难以还原成Fe2+,导致催化活性低、稳定性差,因而实际应用受到限制[11].近年来,多金属催化剂因具有多催化活性位点引起研究者的兴趣.常见的多金属固相催化剂有尖晶石型铁氧体(MN2O4,其中:M、N为金属)[12-14]、铜铁矿型氧化物(MNO2)[15-16]和多金属复合催化剂[17-19].研究人员发现将光催化技术与芬顿技术进行耦合时,光生电子可被Fe3+捕获,将Fe3+还原为Fe2+,促使活性中心Fe2+得以再生,同时光生电子被Fe3+捕获后促进了光生电子和空穴的分离,提高了光催化效率[20].然而,紫外光稳定性差、成本高,其在太阳能光谱中所占比例非常低,仅为4%.因此研发可见光响应的异相光芬顿催化剂至关重要[21-22].铜铁矿 CuFeO2在地球中储量丰富、环境友好且具有较窄的禁带宽度,并展现出良好的光催化和芬顿活性[23],因而在难降解有机污染物处理方面显示出良好的潜力.然而,利用传统的固相烧结法和溶胶-凝胶法制备CuFeO2时需要较高温度或大量使用有机溶剂,不仅制备成本高,还会在反应过程中产生大量杂质.
本文通过低温水热法制备高纯度R-3m型CuFeO2催化剂,并通过各种表征手段对其结构及形貌等进行分析.以OFX作为目标污染物,考察所制CuFeO2催化剂在可见光下活化H2O2降解OFX的效能、机制和循环稳定性.深入探究催化剂投加量、H2O2浓度、pH值对OFX降解效果的影响.利用紫外可见漫反射光谱、X射线光电子能谱分析CuFeO2的禁带宽度和光学性能;通过自由基、光生电子和光生空穴捕获实验和电子自旋共振光谱鉴定可见光/CuFeO2/H2O2体系降解OFX的主要活性物质,并进一步揭示其降解机理.
1 材料与方法
1.1 异相催化剂CuFeO2制备
称取2.4 g的 Cu(NO3)2·3H2O和 4.1 g Fe(NO3)3·9H2O于30 mL去离子水中,室温下磁力搅拌30 min获得完全溶解的黄绿色盐溶液,加入0.5 g无水葡萄糖,待溶解后用0.5 mol·L-1的NaOH调节溶液的pH至11,最后将溶液缓慢倒入100 mL的不锈钢反应釜中,于180℃下水热反应24 h,待黑色产物冷却至室温后分别用去离子水和乙醇对其进行多次清洗,并于60~70℃下干燥,得到CuFeO2催化剂,并将其装袋备用.
1.2 材料的测试表征
采用X-射线多晶衍射仪(XRD,D8 ADVANCE,German)对所制备CuFeO2的晶型结构进行分析,其中测试靶元素为Cu靶,Kα射线的波长为1.54Å,测试范围为10°~90°.采用扫描电子显微镜(SEM,JEOL-7500F,Japan)、能量色散 X射线能谱仪(EDS)以及透射电子显微镜(TEM,Tecnai G2 f20 s-twin)对CuFeO2的表面形貌以及表面尺寸结构特征进行分析.以Mg Kα射线(1 253.6 eV)作为激发源,采用Thermo Fisher Scientific公司所产X射线光电子能谱仪(XPS,ESCALAB 250,American)测定所制备CuFeO2的价带谱.CuFeO2的光吸收性能以及禁带宽度利用美国PE公司的紫外可见光谱仪(UV-Vis/DRS,Lambda 900,American)进行测定.
1.3 异相催化剂CuFeO2可见光下活化H 2 O2降解OFX的实验
OFX的降解实验采用外置光源型反应系统,主要由圆柱形的石英反应器、磁力搅拌器和300 W 的氙灯组成.反应器加入100 mL 10 mg·L-1的OFX溶液和一定剂量的CuFeO2催化剂.光芬顿催化降解反应前,先将反应器置于黑暗处搅拌1 h,达到吸附-解吸平衡,随后开启氙灯和搅拌器启动催化反应.每间隔30 min取样,采用日本岛津仪器公司的L-2000高效液相色谱仪(HPLC)测定OFX的残余浓度,HPLC采用Ultimate Plus-C18型色谱柱,尺寸为4.6 mm×150 mm×0.005 mm,柱温为30℃,流动相为乙腈-水(V:V=60:40),流速为 1.0 mL·min-1,检测波长为293 nm.采用电子自旋共振波谱仪(ESR,FA200)检测降解过程中的活性自由基.分别利用式(1)和式(2)计算OFX的降解效率和拟一级动力学反应速率常数.
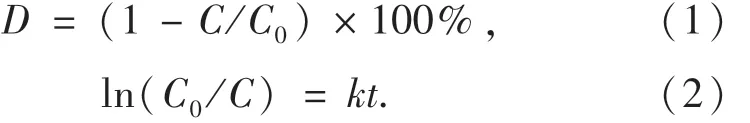
式中:D为OFX的降解效率;C0为OFX的初始浓度,mg·L-1;C为异相光芬顿降解时间为t时OFX的浓度,mg·L-1;k为一级动力学反应速率常数,min-1.
2 结果与讨论
2.1 CuFeO2催化剂的表征
2.1.1 晶型结构和表面形貌
通过XRD、SEM、TEM和HRTEM等手段分析了所制CuFeO2催化剂的晶型结构和表面形貌,如图1所示.由图1(a)显示,经过与 PDF#75-2416标准谱图进行比对,所制催化剂在10°~80°出现的特征峰与R-3m相CuFeO2的峰型一一对应,反映3R型铜铁矿CuFeO2制备成功.由图1(b)的SEM图可知,所制CuFeO2为斜方六面体结构,其表面光滑均匀、棱角分明,粒径在 1~4μm 之间,不同粒度的CuFeO2颗粒团聚在一起,以团簇物的形式存在.图1(c)所示的TEM图也清晰展现了微米级CuFeO2的斜方六面体结构形态和团聚状态.
2.1.2 能带结构
为了分析p型半导体CuFeO2的能带结构,对其紫外可见漫反射光谱和XPS价带谱进行了测定.根据Kubelka-Munk公式(见式(3))绘制曲线如图2所示.由计算可知,所制CuFeO2的禁带宽度Eg值为2.73 eV.由 XPS价带谱(见图3(a))可知,CuFeO2的价带位置为0.89 eV,故可得其导带位置为-1.84 eV,由此画出如图3(b)所示的能带结构图.

图 1 CuFeO2的 XRD图、SEM 图和 TEM 图Fig.1 XRD,SEM and TEM patterns of CuFeO2
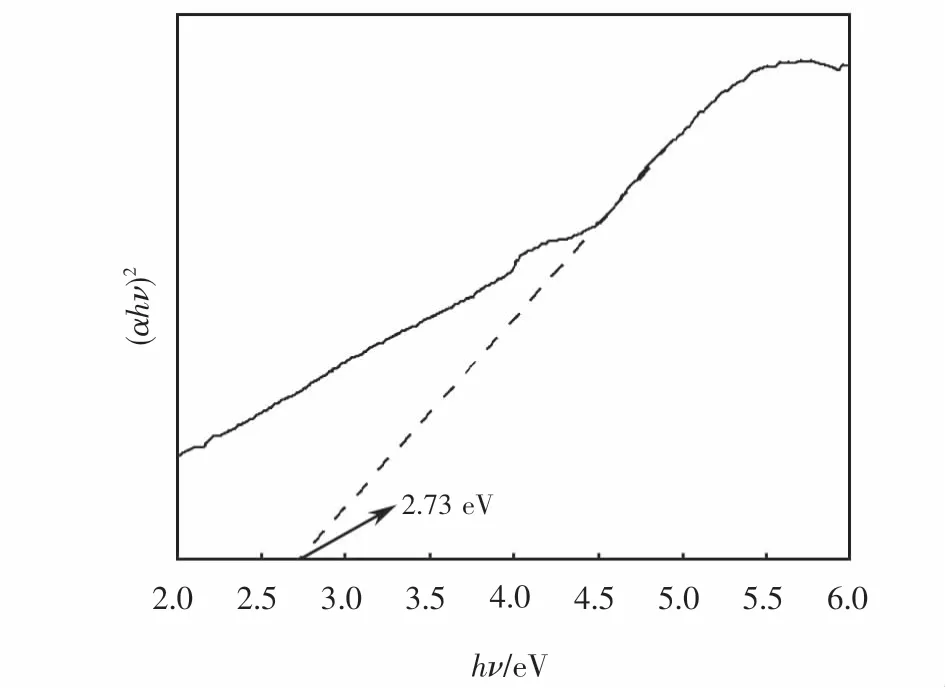
图2 CuFeO2的紫外可见漫反射图Fig.2 UV-Vis DRS of CuFeO2
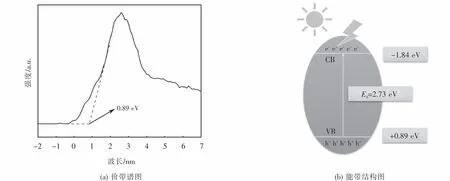
图3 CuFeO2的价带谱和可能的能带结构Fig.3 Valence band XPS spectra and the possible band structures of CuFeO2
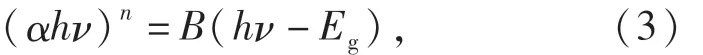
其中:h为普朗克常量;ν为光的频率;B为常数;Eg为带隙能量值;α为吸光度;n为1/2或者2(间接带隙型半导体为1/2,直接带隙型半导体为2).
2.2 可见光/CuFeO2/H 2O2 体系下降解 OFX的研究
2.2.1 不同体系下OFX的降解
为了考察可见光响应下所制CuFeO2活化H2O2对OFX的降解效果,分别在不同体系下进行了降解实验并对相应的动力学行为进行了分析,降解条件如下:[H2O2]0=6 mmol·L-1,[CuFeO2]0=0.6 g·L-1,[OFX]0=10 mg·L-1.如图4(a)所示,在未调节降解溶液初始pH的条件下,反应120 min后,单独用6 mmol·L-1H2O2氧化降解10 mg·L-1OFX时,其去除效率仅为0.58%,这是由于H2O2的氧化还原电位值较低仅为1.77 eV,对有机污染物的氧化能力较弱[24];单独可见光照射污染物时,其降解效率为20.2%,表明单独可见光照射对OFX的去除效果不佳;CuFeO2催化剂对OFX的吸附曲线显示,CuFeO2在降解1 h后达到吸附-解吸平衡,继续延长吸附时间,由于CuFeO2的团聚,减少了CuFeO2颗粒与OFX分子在水中接触的机会,因而OFX的浓度几乎不再改变,CuFeO2对OFX的吸附率仅为19.9%;与单一体系相比,可见光/H2O2和可见光/耦合体系对OFX的降解效率分别增加25.4%和30.1%,这说明可见光对H2O2以及 CuFeO2均有一定的活化能力;当H2O2和CuFeO2耦合作用于污染物时,其降解效率为41.9%,这是因为CuFeO2能够有效地活化H2O2产生自由基,促进OFX地降解;而本文所选可见光/CuFeO2/H2O2体系对OFX的降解效率高达85.3%,其较高的催化降解性能源于可见光和CuFeO2对H2O2的协同催化作用.此外,通过拟一级动力学方程进一步分析了OFX的降解过程.由图4(b)可知,可见光/CuFeO2/H2O2体系降解 OFX的速率常数(1.48×10-2min-1)分别是 H2O2氧化(3.10×10-5min-1)和CuFeO2吸附(1.91×10-3min-1)的774.9倍和7.8倍,进一步说明CuFeO2可以作为一种有效的异相光芬顿催化剂活化H2O2降解OFX.
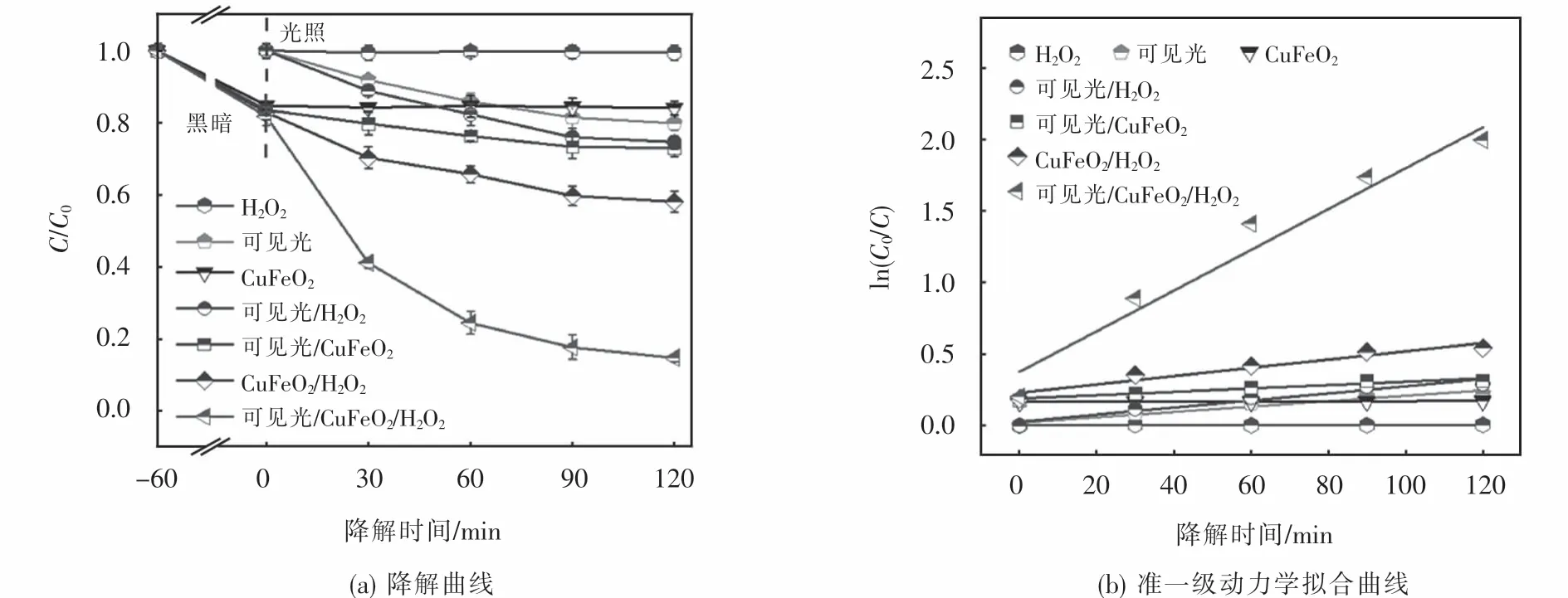
图4 不同体系下OFX的降解曲线和准一级动力学拟合曲线Fig.4 Degradation and corresponding kinetic curves of OFX in various system
2.2.2 催化剂投加量和H2O2浓度对OFX降解的影响
在浓度为 10 mg·L-1的 OFX、10 mmol·L-1H2O2的条件下,通过改变CuFeO2催化剂的投加量,探究其在光芬顿体系下对OFX降解性能的影响,如图5(a)所示,当CuFeO2催化剂的投加量从0.1 g·L-1增至 0.6 g·L-1时,可见光/CuFeO2/H2O2体系对OFX的降解效率由39.3%增至82.6%,这是由于随着催化剂投加量地增加,吸附和催化H2O2的活性位点和光生载流子数目也随之增加,促进了体系对OFX地降解.但是当CuFeO2催化剂投加量进一步增加至0.8 g·L-1时,OFX的去除效率由82.6%降为80.1%.这种现象是因为CuFeO2催化剂投加量地增加虽然会使活性位点和光生载流子数目增加,但受限于H2O2浓度,过量的催化剂活性位点无法得以良好的利用[25].此外,CuFeO2的过量投加还可能会造成溶液中活性自由基的不良消耗以及减弱可见光在体系中的透过性,对OFX的降解产生抑制作用.因此,0.6 g·L-1CuFeO2为异相光芬顿降解OFX的最佳催化剂投加量.
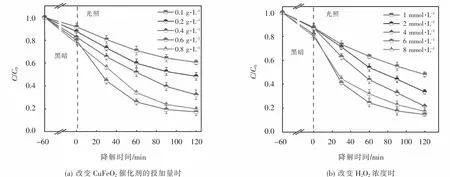
图5 CuFeO2投加量、H 2 O2浓度对OFX降解的影响Fig.5 Effect of CuFeO2 dosage and H 2 O2 concentration on OFX degradation
在最佳CuFeO2催化剂投加量时,继续探究H2O2浓度对可见光/CuFeO2/H2O2体系降解OFX的影响.由图5(b)可知,当 H2O2浓度由1 mmol·L-1增至6 mmol·L-1,OFX的降解效率从56.4%增至85.2%,这是由于随着H2O2浓度增加,体系中会产生更多的活性自由基,加速 OFX地降解.然而,将H2O2浓度进一步提高至8 mmol·L-1时,体系对OFX的去除效率下降至83.4%.上述结果表明,在一定范围内提高H2O2浓度可以增加活性自由基的数量,然而过量的H2O2会通过式(4)和(5)发生副反应导致·OH的不良消耗,产生比·OH氧化能力更低的·O2H和·O-2[24],从而抑制了 OFX地降解.因此,异相光芬顿催化降解OFX的最佳H2O2浓度为6 mmol·L-1.
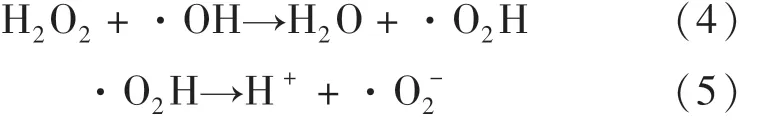
2.2.3 溶液初始pH对OFX降解性能的影响
在CuFeO2投加量为0.6 g·L-1和H2O2浓度为6 mmol·L-1条件下,考察了初始pH对可见光/CuFeO2/H2O2体系降解OFX的影响,其降解条件如下:[OFX]0=10 mg·L-1;[H2O2]0=6 mmol·L-1;[CuFeO2]0=0.6 g·L-1.由图6可知,在未调节溶液初始pH(pH=3.6)时,经过120 min地降解,所制CuFeO2催化剂在可见光下活化H2O2对OFX的降解效率高达85.2%.当溶液pH调节至3,在降解时间为0~60 min时,OFX的降解效率明显高于pH=3.6,这是因为在溶液pH为3时,体系中能够产生足量的H+,使得可见光/CuFeO2/H2O2体系在短时间内产生大量活性自由基,从而促进OFX地降解.随着降解反应地进行,溶液中积累的大量中间产物与OFX地降解产生竞争,导致OFX降解速率下降,其在120 min时的降解效率为85.7%,这与pH=3.6时的降解效率(85.2%)基本一致.然而当调节溶液初始pH为5、7和9时,OFX的降解率分别为76.7%、66.6%和47.4%,这是由于体系中pH的增加会产生高浓度OH-,从而加速了H2O2和活性自由基的不良消耗,使OFX的降解效率下降[26].综上可知,在酸性体系下,CuFeO2催化剂在可见光下活化H2O2对OFX的降解效率最高,随着pH的增加,可见光/CuFeO2/H2O2体系对OFX地降解产生抑制作用,尽管溶液pH为3时比未调节pH时OFX的降解速率更快,但在调节溶液pH过程中需要添加大量的酸,不仅过程繁琐还会增加处理成本,因此,本文其他实验均未调节溶液初始pH.
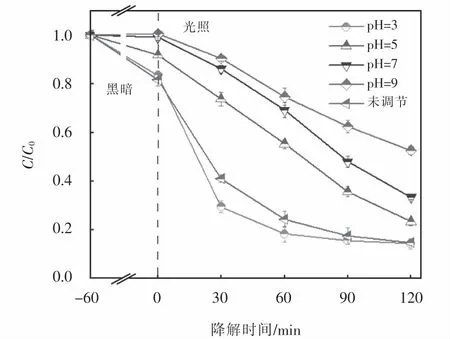
图6 初始pH对OFX降解的影响Fig.6 Effect of initial pH on OFX degradation
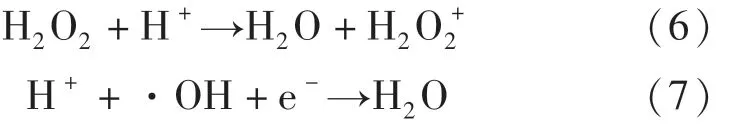
2.3 催化剂循环稳定性分析
为了评价可见光下CuFeO2活化H2O2的稳定性,在CuFeO2添加量为0.6 g·L-1,H2O2浓度为6 mmol·L-1和未调节溶液pH的条件下进行了连续5次催化降解OFX实验.由图7(a)可知,经过5次(连续600 min)的循环降解实验,可见光/CuFeO2/H2O2体系对OFX的降解效率从最初的85.2%降低至77.5%,说明本研究所制CuFeO2作为异相催化剂在可见光下活化H2O2过程中具有良好的稳定性.通过分析5次催化降解反应前后CuFeO2的XRD谱图的晶型结构,进一步探讨了CuFeO2催化剂的稳定性.由图7(b)可知,CuFeO2的XRD谱图经过5次连续循环降解实验几乎没有发生变化,其仍能保持原有晶型结构.上述结果证实,可见光/CuFeO2/H2O2体系降解OFX可以在较长时间内保持较高的催化活性.
图7 可见光/CuFeO2/H 2 O2体系对OFX的5次循环降解,降解前后的XRD图Fig.7 Five successive degradation cycles of CuFeO2/H 2 O2 system for the OFX degradation and XRD patterns before and after the degradation
2.4 CuFeO2催化剂Fenton体系下降解OFX机理
2.4.1 鉴定活性自由基
为了鉴定 CuFeO2可见光下活化 H2O2降解OFX的主要活性自由基,本研究分别采用甲醇、硝酸银、对苯醌和草酸铵作为·OH、光生电子、·O2-和光生空穴的捕获剂,其降解条件如下:[H2O2]0=6 mmol·L-1,[CuFeO2]0=0.6 g·L-1,[OFX]0=10 mg·L-1和[捕获剂]0=50 mmol·L-1.由图 8(a)可知,与不加捕获剂的对照组相比,甲醇、叠氮化钠、硝酸银、对苯醌地添加明显抑制了OFX地降解,而草酸铵地添加则对OFX降解没有明显影响.当添加甲醇、硝酸银、对苯醌后,经过120 min的降解实验,OFX的降解效率从对比组的85.2%分别下降到1.9%、53.2%、58.1%,而草酸铵添加后OFX的降解效率仅下降了3.1%.以上结果表明,·OH、光生电子、·O2-均在可见光/CuFeO2/H2O2体系降解OFX中起作用,而·OH是异相Fenton催化降解OFX的主要活性自由基.为了进一步研究可见光/CuFeO2/H2O2体系降解OFX生成活性自由基的情况,以二甲基吡啶N-氧化物(DMPO)为捕获剂,分别在光催化体系(可见光/CuFeO2)、异相芬顿体系(CuFeO2/H2O2)和异相光芬顿体系(可见光/CuFeO2/H2O2)下进行ESR实验.ESR谱图(见图8(b))显示光催化体系内出现了极微弱的DMPO-·OH和DMPO-O-2信号峰,表明光催化体系无法产生·OH和·O-2,这与降解实验结果相符合.而异相芬顿体系和异相光芬顿体系中均检测到强度比为1:2:2:1的DMPO-·OH加合物的特征峰和强度比为1:1:1:1的DMPO-·O-2加合物的特征峰,表明CuFeO2/H2O2和可见光/CuFeO2/H2O2体系均可以产生·OH和·O-2.然而,在相同反应时间内,以上两个体系DMPO-·OH特征峰的强度比DMPO-·O-2特征峰强度大,表明·OH是异相芬顿和异相光芬顿体系中的主要活性自由基,这与自由基捕获实验的结果一致.此外可见光/CuFeO2/H2O2体系特征峰的强度明显高于CuFeO2/H2O2体系,这说明该体系下能够产生的自由基浓度更高,这与图4降解实验相一致.
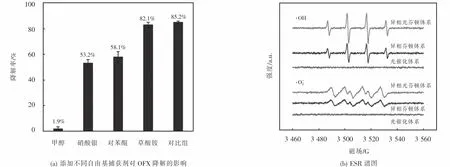
图8 不同自由基捕获剂对OFX降解的影响以及DMPO-·OH和DMPO-·O-2 的ESR谱图Fig.8 Effect of various radical scavengers on OFX degradation and ESR spectra of DMPO-·OH and DMPO-·O-2
2.4.2 CuFeO2催化H2O2降解OFX的机理
基于自由基、光生电子和光生空穴捕获实验以及ESR分析结果,提出了如图9所示CuFeO2可见光下活化H2O2降解OFX的可能机理,整个降解反应主要发生在CuFeO2表面的Cu和Fe活性位点上.当CuFeO2投加至污染物溶液中,一方面催化剂表面的Cu和Fe作为Lewis位点经过式(8)反应生成≡Cu+/Cu2+/Fe3+-OH络合物,其中≡Cu+-OH可以通过式(9)与H2O2反应生成·OH和≡Cu2+-OH,生成的≡Cu2+-OH可经式(10)被H2O2还原成≡Cu+-OH并生成·O-2.与此同时,CuFeO2表面的≡Fe3+-OH也能与H2O2反应生成≡Fe2+-OH和·O-2(式(11)),而CuFeO2催化剂表面形成的≡Fe2+-OH又能通过式(12)催化 H2O2生成·OH 和≡Fe3+-OH.由于Cu2+/Cu+的氧化还原电位为 0.17 V,远远低于Fe3+/Fe2+的氧化还原电位值(0.77 V),因此 CuFeO2表面的≡Cu+-OH能将≡Fe3+-OH还原成≡Fe2+-OH(式(13)).通过催化剂表面的≡Cu+-OH/≡Cu2+-OH和≡Fe2+-OH/≡Fe3+-OH的氧化还原循环可以促进活化H2O2生成活性自由基.另一方面,经过可见光激发,CuFeO2可以通过式(14)产生光生电子和光生空穴.由图3可知,CuFeO2的价带位置为+0.89 eV,这比·OH/OH-的电势(+2.4 eV)或·OH/H2O的电势(+2.72 eV)更负,因此光生空穴不能将H2O氧化成·OH,而被激发产生的光生电子可以直接通过并将H2O2转化成·OH(见式(15)).此外光生电子可以通过式(16)和式(17)促进催化剂表面Cu+和Fe2+的原位再生,极大地促进了活性自由基地产生.最终产生的·OH/·O-2可将OFX氧化成中间产物 CO2和 H2O(见式(18)).
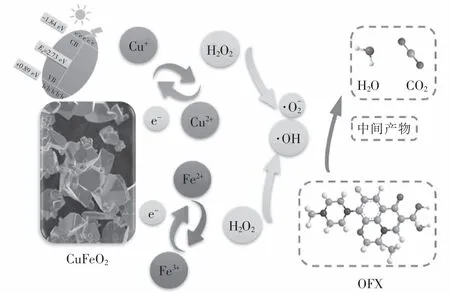
图9 可见光/CuFeO2/H 2 O2体系中催化反应的机理示意图Fig.9 Possible mechanism of OFX degradation by CuFeO2 in visible/CuFeO2/H 2 O2 system
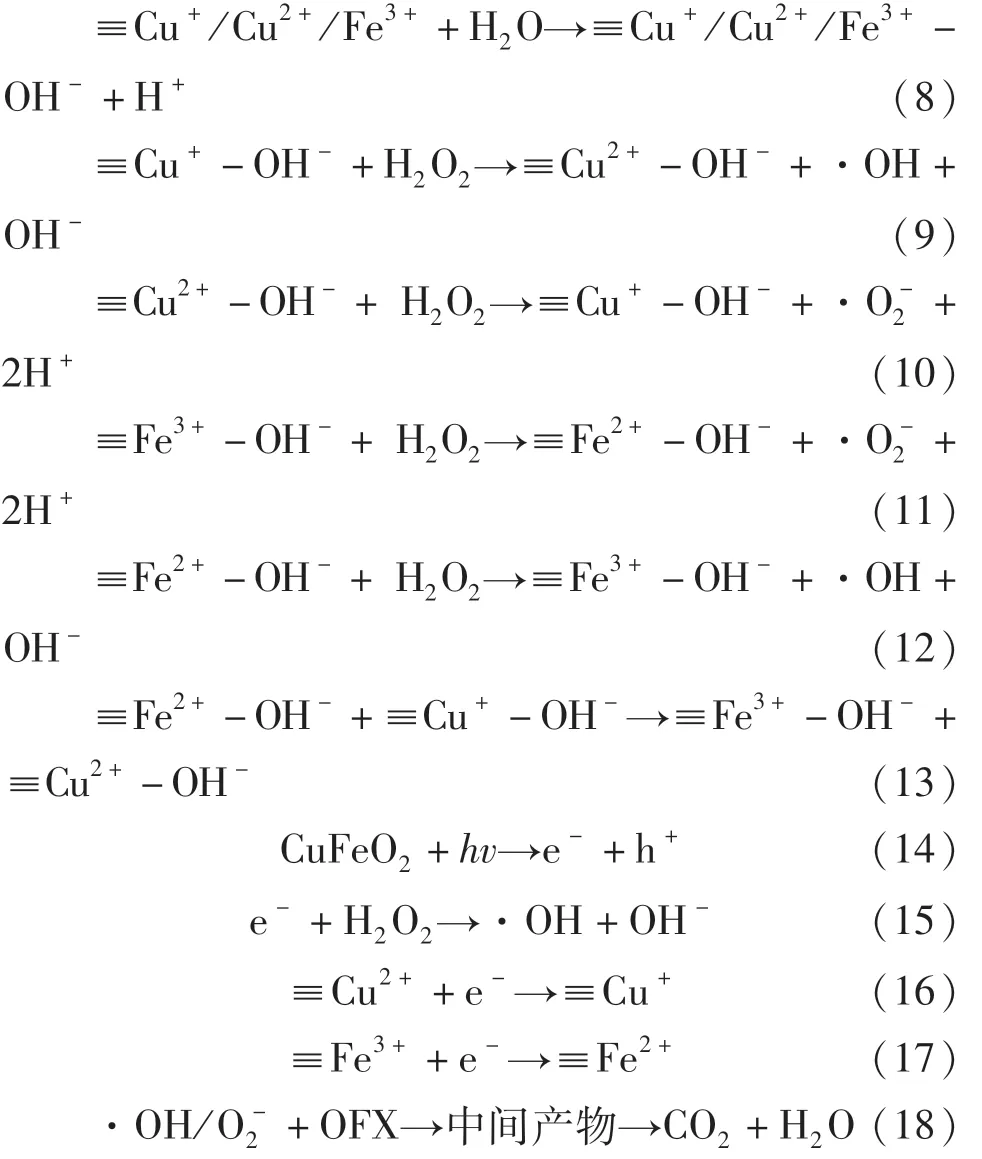
3 结论
1)通过水热法成功制备高纯度R-3m相结构CuFeO2催化剂,其表面光滑均匀、棱角分明,粒径在1~4μm之间,且CuFeO2颗粒呈团聚状态.
2)探究了可见光/CuFeO2/H2O2体系降解OFX的最佳降解参数,即催化剂浓度为0.6 g·L-1,H2O2摩尔浓度为6 mmol·L-1以及pH=3.6.在最佳降解条件下,CuFeO2催化剂在可见光下活化H2O2降解OFX的效率为85.2%.
3)经5次连续降解后,可见光/CuFeO2/H2O2体系降解OFX的效率仅降低了7.7%;XRD谱图证实降解反应后CuFeO2仍能保持原有的晶型结构,结果表明所制CuFeO2具有良好的稳定性.
4)自由基、光生电子和光生空穴捕获实验以及ESR测试结果表明可见光/CuFeO2/H2O2体系中·OH为主要活性自由基;提出了可能的降解机理,揭示了CuFeO2中的Fe和Cu双活性位点能够加速Fe3+/Fe2+和Cu+/Cu2+的氧化还原循环,促进活性自由基地生成,加速OFX地降解.
猜你喜欢
科学之友(2022年11期)2022-11-03
工业安全与环保(2022年10期)2022-10-28
材料与冶金学报(2022年2期)2022-08-10
工业水处理(2022年6期)2022-06-23
石油化工高等学校学报(2021年3期)2021-07-15
云南化工(2020年11期)2021-01-14
作文成功之路·小学版(2020年6期)2020-07-27
浙江大学学报(理学版)(2020年1期)2020-03-12
天津城建大学学报(2015年5期)2015-12-09
安徽农学通报(2015年2期)2015-02-12
