
丁烯/丁醇异构体低温氧化Waddington 反应机制理论研究
2022-10-29 11:58ManiSarathy黄佐华张英佳
燃烧科学与技术 2022年5期
赵 倩,李 阳,Mani Sarathy,黄佐华,张英佳
(1.西安交通大学动力工程多相流国家重点实验室,西安 710049;2.西北工业大学燃烧、结构与内流场重点实验室,西安 710072;3.阿卜杜拉国王科技大学清洁燃烧研究中心,图沃 23955,沙特阿拉伯)
低温燃烧是提高热效率、降低NOx和碳烟排放双重目标的有效手段.对燃料低温化学的准确剖析对其低温燃烧应用十分关键.Waddington 机制在烯烃和醇类的氧化化学反应动力学中的重要性主要体现在两个方面:①传统低温氧化途径的竞争性反应通道;②连接烃类和醇类低温氧化动力学的桥梁.与传统烷烃低温氧化过程不同,烯烃氧化过程中的Waddington 机制中反应第一步的异构化反应涉及的是OH中—OH 位H 原子转移,而不是C 原子上的H 原子转移.此外,OH除了由烯烃与OH 自由基加成得到,还可通过醇类分子的β-位氢提取反应获得.这意味着,Waddington 机制使得烯烃和醇类低温氧化动力学机理相互关联.丁烯作为包含同分异构体的最简单烯烃,已被大量研究证实Waddington 机制在其低温氧化反应动力学过程中的重要性.
国际上较早就已开展有关Waddington 机制的研究.1965 年,Knox[1]在对气相中烃类低温氧化动力学研究中首次提出H2转化为H 自由基的反应新机制,其中,烯烃(A=B)作为大多数烷烃的初级氧化产物,其共氧化必须要包含在该反应新机制中.其主要过程为:
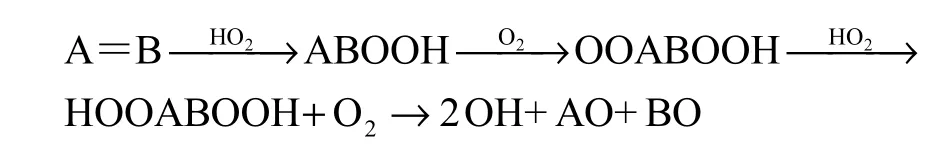
随后,Ray 和Waddington 等[2-5]利用该转化机制解释了2-丁烯氧化中乙醛产率过高的原因,并在乙烯、丙烯和异丁烯的低温(184~400 ℃)氧化反应实验中对该反应机制进行验证.1989 年,Wilk 等[6]在研究丙烯的低温氧化时,正式将其命名为Waddington机制.图1 给出了烯烃氧化过程中的Waddington 反应历程(其中,R1~R4 表示任意的烷基或氢原子),包括:①导致C=C 双键打开的烯烃H 自由基加成,形成β烷基过氧化氢自由基OH(β-alkyl-hydroxy radical);②OH的氧气加成,产生烷基氢过氧化物R(O)OH(alkyl-hydroxy-peroxy radical),经过六元环过渡态,发生羟基位的 H 迁移,异构化形成ROOH(alkyl-hydroperoxide-alkoxy radical) ;③ROOH上C—C 键和O—O 键断裂,形成甲醛/酮和H 自由基.这一反应过程中,经六元环过渡态结构的异构化以及后续解离过程(蓝色部分)被称为典型的Waddington 机制.与传统烷烃低温氧化过程不同的是,Waddington 机制中反应第一步的异构化反应涉及的是—OH 位的H 原子转移,而不是C 原子上的H 原子转移.值得注意的是,OH除了由烯烃与H 自由基加成得到,还可通过醇类分子的β-位氢提取反应获得,这意味着,Waddington 机制使得烯烃和醇类低温氧化动力学机理相互关联.

图1 烯烃氧化的Waddington机制Fig.1 Waddington mechanism in alkene oxidation
通过文献回顾[7-9],以及查阅最近有关丁醇异构体[10]、异丁烯[11]、2-丁烯[12]和1-丁烯[13]详细反应机理研究中发现,Waddington 机制在烯烃和醇类的氧化化学反应动力学中十分关键.Li 等[13]在对1-丁烯动力学模型的低温氧化路径分析时发现,在2 MPa、700 K 下,约 27% 的 乙 烯 与H 加 成 产 生OH(CH3CH2HCH2OH);形成的OH随后与O2加成生成R(O)OH(CH3CH2CH(O)CH2OH),其中约26.5%会经六元环过渡态异构化后裂解产生乙醛、甲醛和H 自由基.Waddington 反应通道较大的分支占比足以体现出其对丁烯低温氧化的重要性.图2 为部分1-丁烯低温氧化路径(以20%燃料消耗计).
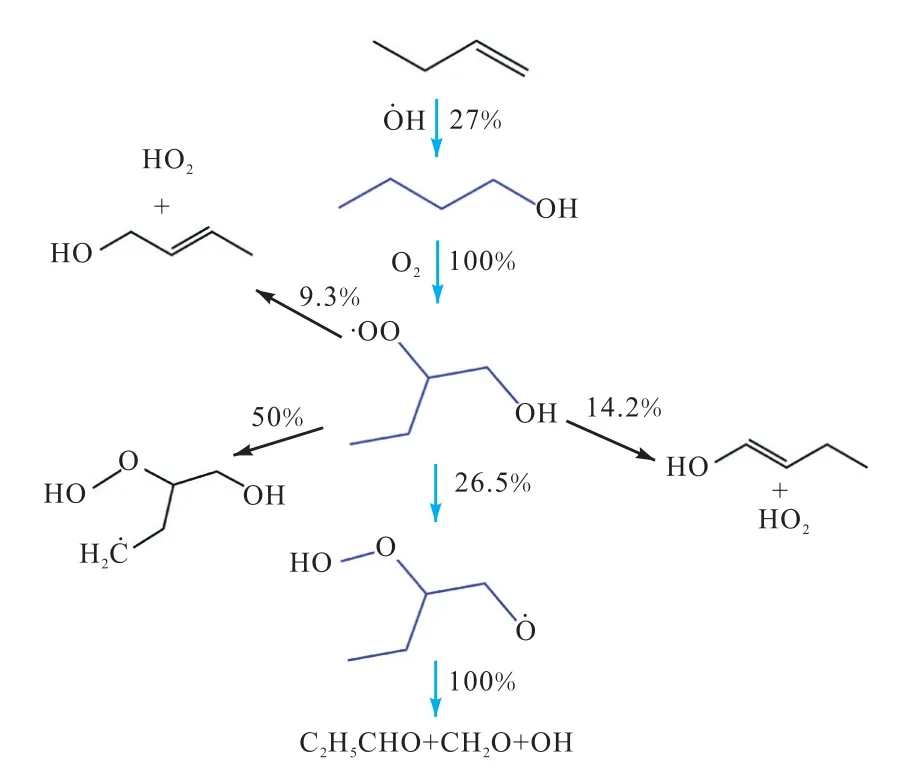
图2 部分1-丁烯低温氧化路径Fig.2 Partial pathways of 1-butene low-temperature oxidation
Welz 等[14]在对异戊醇(3-methylbutan-1-ol)低温(550~750 K)燃烧机理研究中发现,β-RO2(βhydroxyalkylperox,CH2(O)CH2OH)的一个重要反应途径是通过Waddington 机制形成异丁醛、甲醛和H 自由基.并且,Welz 等[15]随后在异丁醇/叔丁醇体系低温(550~700 K)燃烧机理研究中也得到同样的结论.
为了更好地预测Waddington 机制涉及的基元反应速率系数,Boudart[16]提出了一种经验方法来估计异构化反应活化能,具体包括:①类似自由基从分子中提取氢原子的反应活化能;②环状过渡态的形变能.但以这种经验依赖性的方法获取Waddington 机制反应速率系数的可靠性尚未可知.Santiago 和Albert[17]在CCSD(T)/6-311+G(3 df,2 p)//B3LYP/6-31G(d,p)理论水平下计算了乙烯的H 自由基加成以形成H2CH2OH,再与氧气加成形成过氧化物的反应势能面,但未提供直接理论计算的速率系数.Sun 等[18]在CBS-Q//B3LYP/6-31G(d,p)理论水平下对异丁烯氧化过程中的Waddington 机制进行了研究,给出了相关物种热力学和动力学参数.Chen等[19]在CBS-q/MP2(full)/6-31G(d)理论水平下计算了异丁烯的H 加成产物(isobutene-OH adducts),与氧气加成的反应势能面,并基于QRRK 理论提供了异构化反应速率系数.Lizardo-Huerta 等[20]在CBSQB3//B3LYP/6-311G(d,p)理论水平下研究了含β-HORO和HOQOOH 自由基的单分子反应势垒,同样基于QRRK 理论计算了异丁烯和1-丁烯体系中相关反应速率系数.Gabriel 等[21]在 M05/aug-ccpVTZ//B3LYP/6-31G(2 df,p)理论水平和CBS-QB3方法下分别对异戊二烯的Waddington 反应的热/动力学参数进行了计算,但只获得了250~350 K 温度范围内的反应速率系数,而实际燃烧工况下的速率并未给出.Welz 等[22]在CBS-QB3 水平和RQCICD(T)/CBS//B3LYP/6-311G(d,p)水平下构建了正丁醇β-自由基(1-hydroxybut-2-yl,CH3CH2HCH3OH)的氧气加成反应势能面,但也未给出相关反应速率系数.
可以看出,目前还缺乏系统性的、高精度的Waddington 反应机制速率系数理论研究,且鲜有研究关注烯烃/醇同分异构体在微观反应层面对其低温氧化过程的影响.鉴于丁烯是包含同分异构体的最简单烯烃,且已有大量研究证实了Waddington 机制在丁烯低温氧化反应动力学过程中的重要性.鉴于此,本文选取丁烯/丁醇及其异构体为研究对象,基于更高精度的理论计算方法,系统开展其Waddington动力学机制理论研究,以提供该类型反应的热/动力学参数,并对计算结果进行了多方面、多维度的比较.为烯烃、醇类低温氧化反应动力学理论建模提供数据支撑,还可作为长链烯烃和醇类氧化过程Waddington 机制基元反应速率系数的评估准则.
1 理论方法
在M06-2X/6-311++G(d,p)理论水平上确定了相关组分的几何结构,以此为基础,进行了振动分析和零点能计算;分别采用0.983 和0.969 8[23]作为校正因子对理论方法计算误差以及非谐性效应、零点能进行校正.对于优化后的过渡态结构,进行内禀反应坐标扫描,以确保给定过渡态结构能连接到预期的反应物和产物.采用一维受阻转子近似处理—CH3基团、—OH 基团以及—COOH 基团的转振模式,并在M06-2X/6-311++G(d,p)理论水平上评估其转动势垒.上述工作均在Gaussian 09[24]上完成.
平衡计算成本和计算要求的近CCSD(T)理论[25]精度,选择DLPNO-CCSD(T)方法[26]结合cc-pVTZ和cc-pVQZ[27]基组,在ORCA 4.0.0[28]程序包中完成电子能量的计算,并将其外推到完备基组极限:
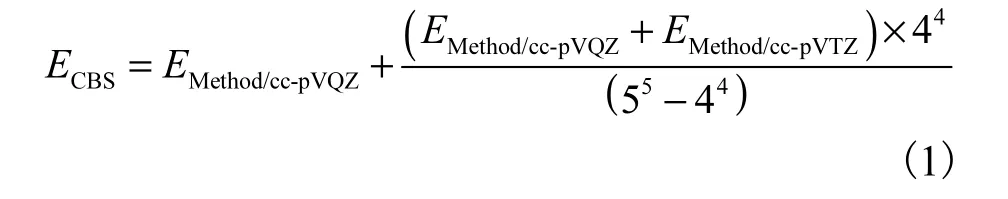
式中:ECBS为外推到完备基组极限的能量;EMethod/cc-pVQZ为CCSD(T)/cc-pVQZ 和DLPNO-CCSD(T)/cc-pVQZ 水平下计算得到的能量;EMethod/cc-pVTZ为CCSD(T)/cc-pVTZ 和 DLPNO-CCSD(T)/cc-pVTZ水平下计算得到的能量.
需要说明的是,DLPNO-CCSD(T)方法有3 个主要截断阈值,分别为 TightPNO、NormalPNO 和LoosePNO,分别在 1.000 kJ/mol、4.186 kJ/mol 和8.372~12.558 kJ/mol 误差内接近CCSD(T)的能量精度.尽管TightPNO 的计算成本较NormalPNO 高出2~4 倍,但比标准CCSD(T)低数量级.因此,本文采用TightPNO 作为DLPNO-CCSD(T)方法的截断阈值.
基于RRKM/ME[29]方法,采用过渡态理论[30],在298.15~2 000 K 温度范围和0.001~10 MPa 的压力范围下求解一维主方程获得目标反应温度和压力依赖的速率系数.使用Eckart 模型[31]评估量子遂穿效应对速率系数的贡献;采用单指数下降模型表征碰撞能量传递概率.由于能量传递参数对温度和尺寸的弱依赖性,对目标反应势能面采用<ΔEdown>为:200×(T/300)0.75cm-1.体系HCH2OH,,CH3C(OH)HC和(CH3)2C(OH)CH2O)的碰撞频率使用Lennard-Jones(LJ)参数评估,本文目标体系类的LJ 参数采用PAPR[32]中的OneDMin 模块计算:σ=0.505 nm,ε=1 449 cm-1;以氮气作为惰性载气,其LJ 参数为:σ=0.361 nm,ε=68 cm-1.利用MultiWell[33-34]计算高压速率极限(HPL:high-pressure limit);使用PAPR[32]计算温度和压力依赖的反应速率系数.计算得到的速率系数以修正阿伦尼乌斯关系式拟合:
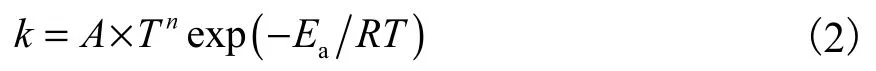
以ATcT 数据库[35]为基准,采用3 种热力学组合方法G3/G4/CBS-APNO[36-38]计算获得目标反应体系相关组分0 K 时的能量,其误差最大不超过4 kJ/mol.在此基础上,使用原子化焓法计算目标反应相关组分标准生成焓:
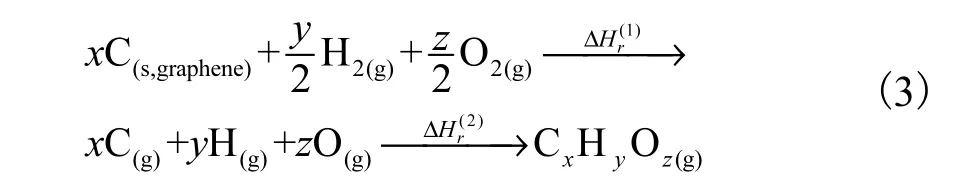
式中:第1 步的反应焓涉及的3C、2H、3O 的标准生成焓参考 ATcT 数据库[35,39-40],分别取值为:716.87 kJ/mol、217.99 kJ/mol 和249.23 kJ/mol.第2步为CxHyOz原子化焓的负值,直接通量子化学方法G3/G4/CBS-ANPO 在Gaussian 09[24]下完成.热力学参数在Multi Well[33-34]和PAPR[32]下获得,并拟合成15 参数的NASA 多项式.
2 结果与讨论
2.1 C4 H 93反应势能面
图3 为0 K 时DLPNO-CCSD(T)/CBS 理论水平下构建的反应势能面,共包含15个物种和10个过渡态结构.R1~R5均可由丁烯加成、O2加成获得,或由丁醇O2加成获得.R1 和R2 表示1-丁烯反应体系,其区别在于H 自由基的加成位置差异,分别对应和R4 和R5 表示异丁烯反应体系;而R3 表示2-丁烯反应体系.势能面上所有驻点的耦合簇T1 诊断值均小于0.03,说明采用单参考方法计算的合理性.
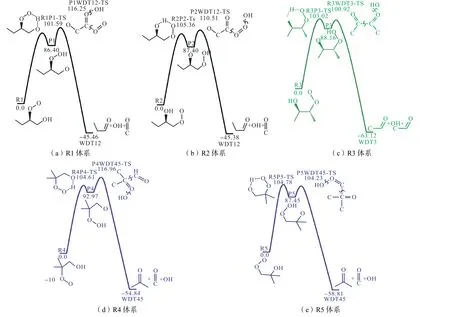
图3 C4 H93反应势能面(单位:kJ/mol)Fig.3 C4 H93 potential energy surface(unit:kJ/mol)
为获得更高精度的能量,本文在CCSD(T)/ccpVXZ//M06-2X/6-311++G(d,p)(X=T,Q)理论水平上计算了上述反应势能面的能垒.并将Sun 等[18]、Chen 等[19]、Lizardo-Huerta 等[20]和Welz 等[22]的研究结果与本文两种理论水平下获得的计算结果进行了比较.由于各研究理论方法的不同,导致计算获得的反应势垒间存在差异,见表1.就Sun 等[18]和Chen等[19]的计算结果而言,尽管其单点能计算均基于CBS-q 理论,但所得结构相差20.93 kJ/mol,误差可能来源于零点能计算及其修正.Welz 等[22]分别基于RQCISD(T)/cc-pVXZ//B3LYP/6-311++G**(X=D,Z)以及CBS-QB3//B3LYP/6-311G**两种理论水平对反应R1↔P1 势垒高度的计算.由于RQCISD(T)方法计算得到的过渡态结构T1 诊断值为0.096,导致其所用单参考方法的能量计算存在较大不确定性,故表1 中仅提供CBS-QB3 理论下得出的反应势垒.基于CCSD(T)/CBS 理论计算得到的异构化反应(R↔P)势垒较DLPNO-CCSD(T)/CBS 理论水平低8.372 kJ/mol,但两种方法得到的解离反应(P↔WDT)势垒接近.
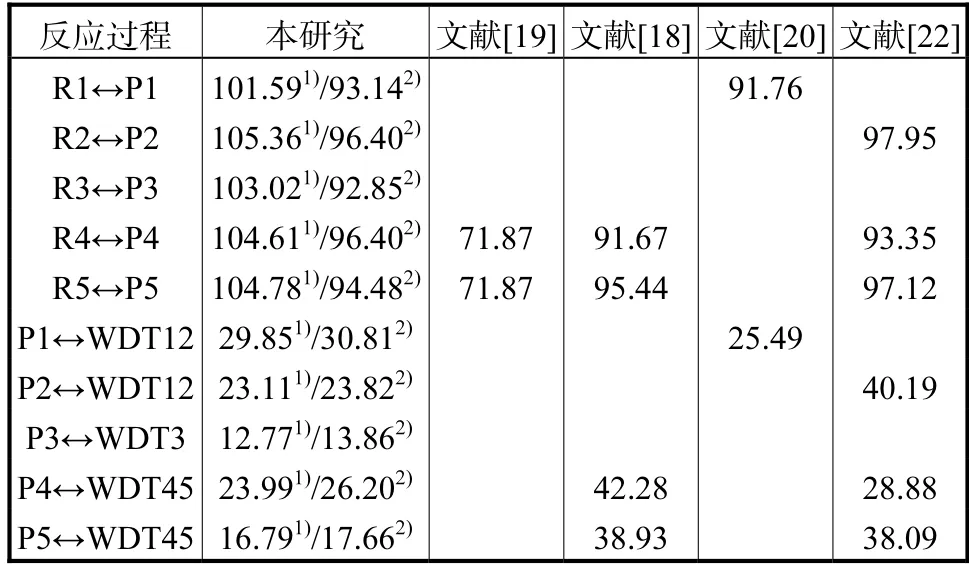
表1 反应势能面上异构化和解离反应的计算势垒高度与文献值对比Tab.1 Comparison of barrier heights for isomerization and dissociation reactions with literature values(kJ/mol)
2.2 速率系数的比较
本文通过多个方面、多个维度对目标体系所有反应速率系数进行了比较,包括基于两种ab-initio 理论计算水平(Gaussian 计算的CCSD(T)和ORCA 计算的DLPNO-CCSD(T));两个不同的动力学参数求解器(MultiWell 和PAPR);不同压力、不同体系的反应势能面(R1、R2、R3、R4 和R5)以及不同反应类(异构化反应、解离反应),总结以下两个方案:①采用两个不同的动力学求解器,分别计算异构化反应(R↔P)和解离反应(P↔WDT)的高压极限速率系数;②基于两种ab-initio 理论得到的反应势垒,系统地计算5 个反应势能面所有反应的高压极限速率系数,并与文献结果进行比较.
2.2.1 基于不同动力学求解器的速率系数比较
基于CCSD(T)/CBS 理论方法获得的能量,使用Multi Well 和PAPR 分别计算了异构化反应和解离反应速率系数,如图4 所示.可以看出,经PAPR 计算的反应速率系数均快于Multi Well 计算结果.对于异构化反应,Multi Well 和PAPR 计算的R1↔P1 反应速率系数差异较明显,但误差不超过32%.在整个计算温度范围内,5 个异构化反应的计算速率系数一致性较好.对于解离反应,5 个体系的速率系数在高温区收敛;且随温度的升高,速率系数对温度依赖性减弱.除R1 体系(P1↔WDT12 反应)以外,两种动力学求解器获得的结果呈现较好的一致性;对于P1↔WDT12 反应,MultiWell 和PAPR 计算的速率系数出现1~2 个数量级差异;温度越高,差异越大.
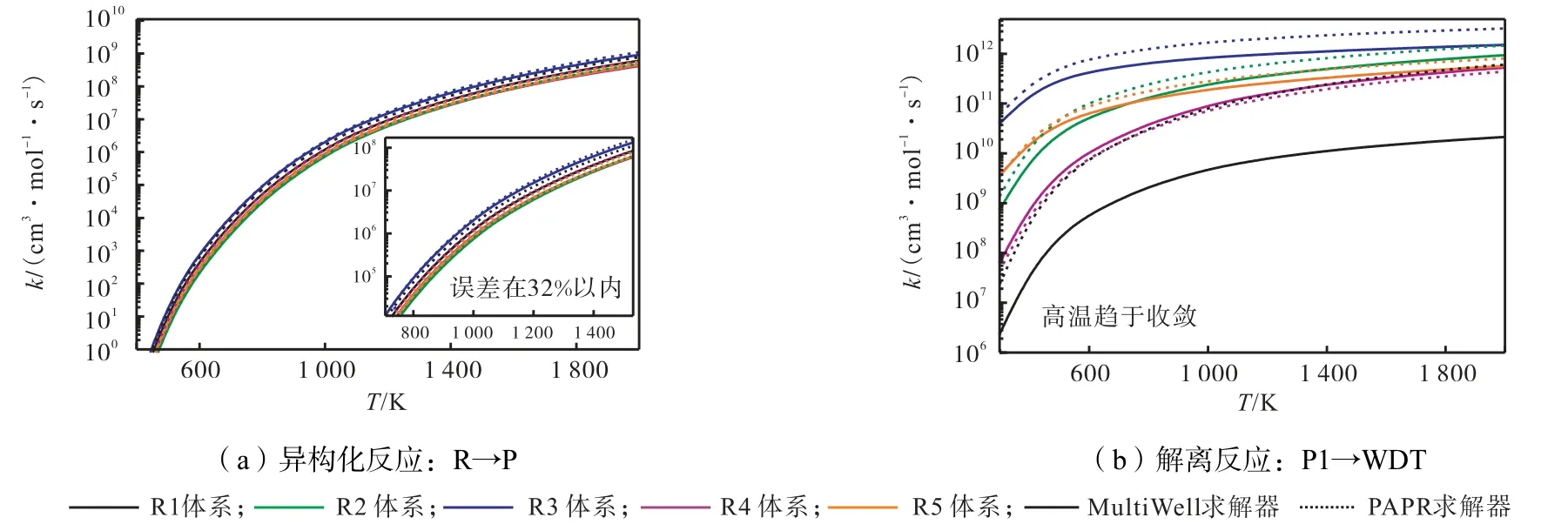
图4 不同动力学求解器计算的异构化和解离反应、高压极限速系数比较Fig.4 Kinetic solver comparison for HPL rate coefficient of isomerization and dissociation reaction
Li 等[41]在1-3 丁二烯的H˙原子氢提取反应和H7势能面相关反应的理论研究中,首次比较了MultiWell 和PAPR 计算的HPL.在其研究结果中,同样出现了两种求解器计算的HPL 存在差异的现象.在势能面各驻点几何结构、相对能量和频率等参数一致的情况下,需进一步分析不同动力学求解器对低频扭转模式势能面的拟合方法及其对配分函数的影响,同时还应考察不同动力学求解器中遂穿系数的评估方法,以便更深入了解不同动力学求解器之间差异.
2.2.2 基于不同量子化学理论方法的速率系数比较
根据3.2.1 小节的比较结果,笔者采用MultiWell对目标体系计算的速率系数进行比较,对于R4 和R5 体系,Sun 等[18]和Lizardo-Huerta 等[20]的研究结果也纳入比较,如图5 所示.对于每个反应势能面,均可观察到解离反应快于异构化反应,差异达1~6个数量级.对于不同反应物,在不同温度下,基于不同ab-initio 理论计算得到的速率系数差异不同.随着温度的升高,差异逐渐减小趋于收敛.意味着,速率系数差异主要来自能量(势垒高度)的差异,而指前因子(振动频率和配分函数)基本保持一致.与CCSD(T)/CBS 理论水平相比,DLPNO-CCSD(T)/CBS 水平计算所得的异构化反应系数较低,而解离反应速率系数较高,主要原因来自其预测的势垒高度,如表1所示.此外,与文献结果相比,本文计算的解离反应速率系数介于Sun 等[18]和Lizardo-Huerta 等[20]的研究结果之间;而本文计算的异构化反应速率系数与文献结果一致,尤其是R5↔P5 反应.
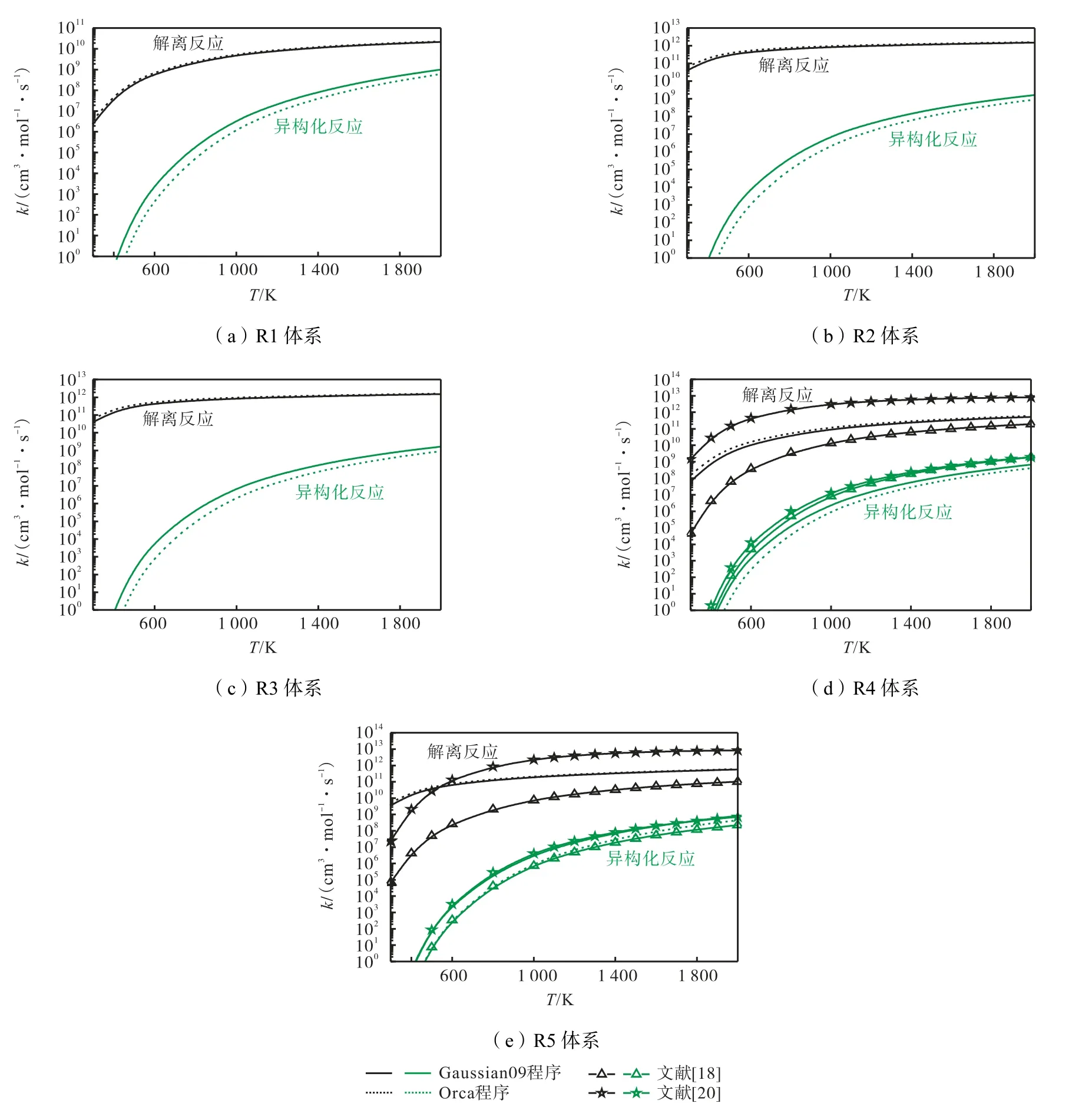
图5 基于不同从头算法能量的异构化和解离反应高压极限速率系数比较Fig.5 Comparison of HPL rate coefficient of isomerization and dissociation reaction based on different ab-initio energies
2.2.3 热力学数据的计算
采用G3、G4 和CBS-APNO 3 种热力学组合方法[36-38],结合原子化焓理论计算了R1~R5 体系所有组分的标准生成焓.基于Somers 和Simme 的研究结论[35],本文最终选取G3/G4/CBS-APNO 等3 种方法计算结果平均值作为标准生成焓.利用Multi Well 计算了所有自由基的热力学参数.对于R4 和R5 体系,本文也利用PAPR 计算了相关组分热力学数据,同时与Sun 等[18]的结果进行了比较,结果见表2.可见,无论是基于MultiWell 计算的热力学数据,还是基于PAPR,均与Sun 等人的结果较好吻合.
表2 C4 H93系列自由基的热力学参数对比Tab.2 Thermochemistry comparison of all C4 H93 radicals
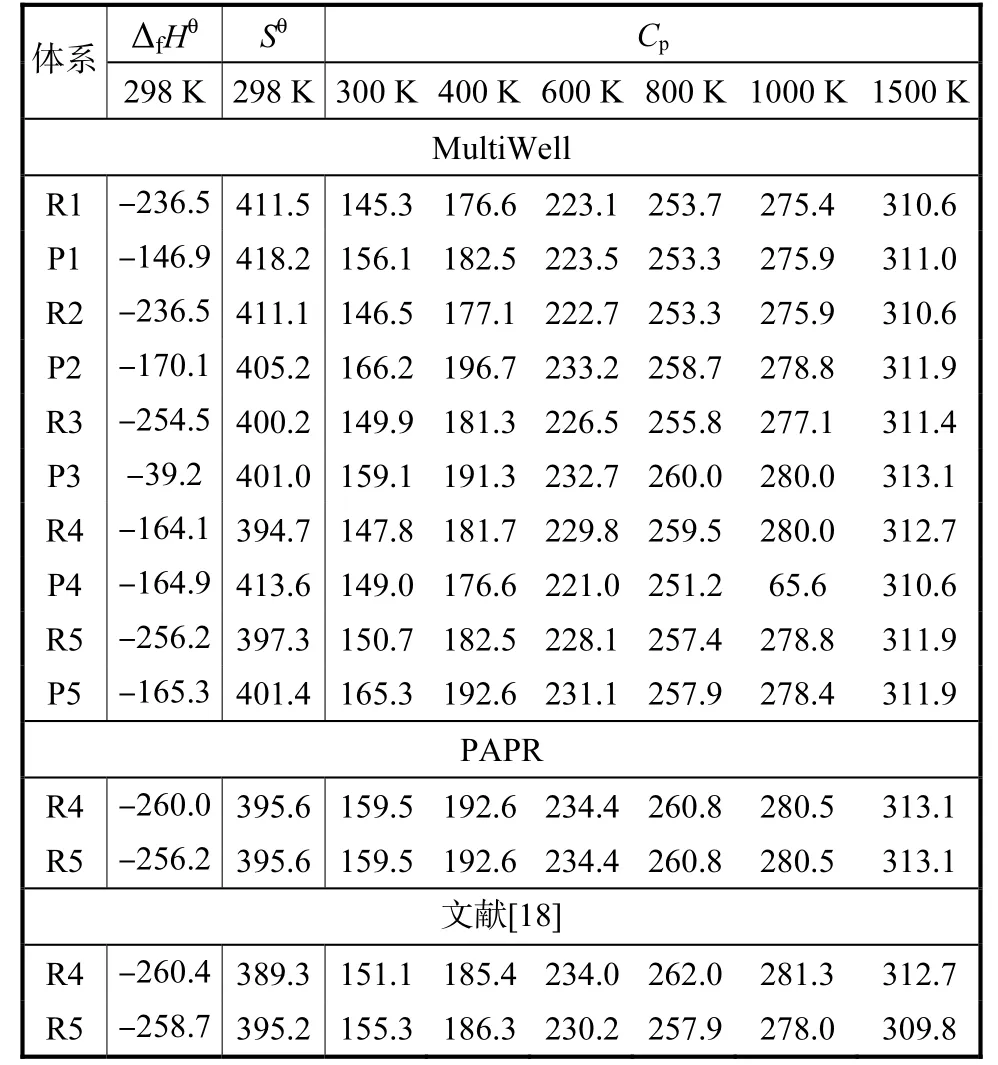
表2 C4 H93系列自由基的热力学参数对比Tab.2 Thermochemistry comparison of all C4 H93 radicals
注:单位:ΔfHӨ:kJ/mol;SӨ:J/(K·mol);Cp:J/(K·mol).
2.3 Waddinton机制对动力学模型性能的影响
精确的速率系数和热力学参数对发展详细化学反应动力学模型十分关键.本文将基于CCSD(T)/CBS 水平得到的Waddington 机制相关反应动力学(HPL)和热力学参数纳入AramcoMech 2.0 模型[12]中查看其对模型预测性能的影响.在原始AramcoMech 2.0 中,Waddington 机制反应速率系数采用Sun 等[18]的推荐值.为了直观比较新计算数据对模型的影响,对化学计量比混合气在3 MPa、650~1 300 K 工况下,进行了丁烯同分异构体着火延迟时间的模拟,详细比较如图6 所示.可以看出,中低温范围(600~900 K),模型修改前后预测着火延迟期差异显著;温度为750 K 时,对异丁烯的预测结果差异达2 倍.
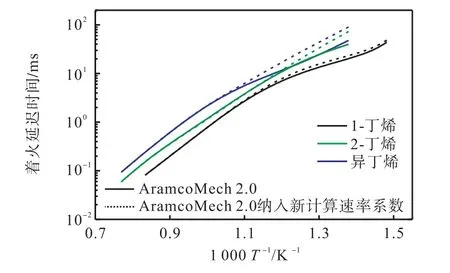
图6 计算Waddington反应速率系数对丁烯异构体着火延迟的影响Fig.6 The effect of calculated Waddington reactionrate coefficient on butane isomer ignition delay
3 结论
本文对丁烯同分异构体和丁醇同分异构体低温Waddington 反应过程开展了系统性理论研究.包括:利用不同ab-initio 理论方法获得了反应势垒,选择MultiWell 和PAPR 两种动力学求解器求解一维主方程获得了异构化反应、解离反应的温度、压力依赖速率系数;采用过渡态理论计算各反应的高压极限.此外,利用Multi Well 和PAPR,基于统计热力学,结合G3/G4/CBS-APNO 方法与原子化焓法计算了所有自由基热力学参数,对计算结果进行了多方面、多维度的比较.
(1) CCSD(T)/CBS 和DLPNO-CCSD(T)理论水平下获得的5 个异构化反应和5 个解离反应势垒高度差异分别为8.372 J/mol 和2.093 kJ/mol,导致基于这两种理论水平势垒计算得出的解离反应速率系数相差较小(小于50%),但异构化反应差异较大(2~10 倍).观察到解离反应速率系数明显快于异构化,且这两种反应速率系数均趋向高温区域收敛.
(2) 两种不同的动力学求解器计算所得的异构化反应速率系数差异很小(小于32%),但对于P1↔WDT12 反应过程,计算速率系数存在1~2 个数量级差异.本文计算的R4 和R5 体系速率系数与文献值吻合较好.此外,本文计算的自由基热力学参数也与文献值吻合较好.
(3) 从动力学建模角度来看,本文计算结果可用于发展丁烯和丁醇异构体中低温燃烧反应机理.此外,还可基于本文计算结果,构建和评估长链烯烃和醇类氧化过程Waddington 机制速率准则.
猜你喜欢
精细石油化工(2022年1期)2023-01-16
——《势能》
文化纵横(2022年3期)2022-09-07
中学生数理化·八年级物理人教版(2022年6期)2022-06-05
中学生数理化·八年级物理人教版(2021年6期)2021-11-22
石油石化绿色低碳(2020年2期)2020-12-31
中学生数理化·八年级物理人教版(2019年6期)2019-06-25
石油石化绿色低碳(2019年6期)2019-01-14
中国塑料(2016年6期)2016-06-27
华东理工大学学报(自然科学版)(2015年3期)2015-11-07
物理化学学报(2015年5期)2015-02-28
