
大麦种子萌发lncRNA-miRNA-mRNA差异表达分析及ceRNA调控网络构建
2022-10-29 01:45张文英高浩然潘锐常浩雯徐乐
长江大学学报(自科版) 2022年6期
张文英, 高浩然, 潘锐, 常浩雯, 徐乐
长江大学作物抗逆技术研究中心,长江大学农学院,湖北 荆州 434025
大麦(HordeumvulgareL.)是世界上最古老的禾本科作物之一,是全球第四大粮食作物,主要用作粮食、饲料加工、啤酒酿造等[1]。种子萌发是植物生命的起始阶段,决定着幼苗的形成和生长[2]。大麦种子的萌发速度和整齐度直接影响啤酒酿造的成本和质量[3]。因此,深入了解大麦种子萌发的分子调控机理意义重大。
近些年,以RNA-Seq为代表的转录组测序技术极大推动了在RNA水平研究基因表达及转录调控机制的研究。在大麦种子萌发方面,mRNA分析揭示了大麦种子萌发过程的耐盐分子机制[4]、赤霉素合成位置以及合成降解途径[5],miRNA分析发现大麦种子萌发过程中miR156等5种miRNA在种胚中高度表达[6]。
长链非编码RNA(long non-coding RNA, lncRNA)是一类本身不编码蛋白、转录本长度超过200nt的长链非编码RNA分子。lncRNA曾被认为是“转录噪音”不具有任何生物学功能。近些年的研究表明,lncRNA在植物的生长发育、表观遗传、胁迫响应等方面起着非常重要的调节功能[7, 8]。
通常情况下,大部分的转录组研究仅关注某一类或两类RNA,没有关注多种类型RNA之间的相互作用及其在转录调控中的作用机制。2011年PANDOLFI 研究小组提出竞争性内源性RNA假说,认为mRNA、lncRNA、环状RNA(circRNA)和假基因转录物通过与miRNA竞争性结合来调节靶基因的稳定性或翻译活性,从而实现转录后功能基因调控。ceRNA机制将miRNA作为相互调节RNA的载体,使编码和非编码基因成为整个转录组内复杂调控网络的有机整体[9]。越来越多的研究表明,ceRNA在动植物的生长发育过程中发挥着重要的调控作用。在玉米种子发育研究中发现了7种新的lncRNA具有潜在的ceRNA功能[10]。利用ceRNA调控网络揭示circRNA在拟南芥叶片发育和水稻旗叶衰老过程发挥着重要作用[11,12]。利用ceRNA调控网络找到6个参与玉米花药发育的靶基因对以及2个参与油菜花药发育的重要circRNA[13,14]。
目前,关于大麦种子萌发过程中lncRNA、mRNA及miRNA构建的ceRNA调控网络尚未见报道。本研究利用大麦种子萌发相关的转录组数据,确定了种子萌发过程中miRNA、mRNA和lncRNA的差异表达谱,筛选种子萌发相关的特异性模块进行GO富集和KEGG分析,构建lncRNA-miRNA-mRNA相互作用的ceRNA调控网络,以期为研究作物种子萌发分子机制提供新思路。
1 材料与方法
1.1 数据来源
从BioProject数据库下载大麦种子萌发相关的RNA-Seq原始转录本数据PRJNA577343和PRJNA578897,其中包括3个品种的36个萌发样本和10个未萌发样本;下载PRJNA389146数据集的miRNA的count数据,其中包括1个种子发育时期和2个种子萌发时期的样本。本研究将种子发育时期视为未萌发种子。
大麦参考基因组的IBSC_v2,基因组DNA序列以及gtf注释文件均从Ensembl Plants下载[15]。
1.2 lncRNA鉴定
采用FASTQC对RNA原始读段进行质量评估和筛选[16]。利用Hisat 2软件将有效读段比对到大麦的参考基因组,计算每个基因的TMM值(Trimmed Mean of M-values)[17]。
采用如下方法鉴定大麦种子萌发相关的lncRNA:利用StringTie软件将每个文库比对结果组装起来,获得转录本结构,利用gffcompare软件将所有文库组装结果合并,剔除与蛋白编码基因重叠的转录本;剔除序列长度小于200nt或含有大于300nt开放阅读框的转录本;将剩余的转录本在Pfam数据库中进行比对,剔除包含保守结构域或者模体的转录本;将剩余的转录本在NCBI-NR数据库进行BLASTx比对,剔除与数据库中蛋白序列同源的转录本;采用CPC2软件评估剩余转录本编码潜能,剔除具有较高编码潜能的转录本;利用Rfam数据库剔除rRNA、tRNA、snoRNA和snRNA等持家非编码RNA[18]。
1.3 差异表达基因筛选
使用DESeq2软件包对mRNA和lncRNA数据进行整理,筛选并确定差异表达的mRNA和lncRNA;利用edgeR函数包对miRNA整理分析,筛选并确定差异表达的miRNA。计算3种不同RNA差异表达倍数,筛选P<0.05且差异表达倍数不低于2倍的转录本为差异表达转录本。
1.4 WGCNA分析
利用R软件WGCNA包分别对差异表达的mRNA和lncRNA构建基因共表达网络分析。根据scalefree拓扑标准,确定合适的β值作为构建网络的软阈值,通过计算所有基因之间的皮尔逊相关性来构建邻接矩阵,并转化为拓扑矩阵,并根据相似性对基因进行层次聚类,挑选连接度高于0.6的特定模块进行分析[19]。
1.5 富集分析及代谢通路分析
利用agriGOv2(http://systemsbiology.cau.edu.cn/agriGOv2/)工具进行GO富集分析,使用基迪奥在线网站平台(https://www.omicshare.com/tools/home/report/koenrich.html)进行KEGG通路分析,根据筛选条件(P<0.05)对分析结果进行汇总,结果以气泡图呈现。
1.6 ceRNA网络构建
利用psRNATarget在线工具对差异表达的miRNA和筛选出的mRNA及lncRNA进行靶基因预测,获得lncRNA-miRNA-mRNA的ceRNA网络数据,利用Cytescape V3.6.1软件进行可视化分析,根据“高表达lncRNA-低表达miRNA-高表达mRNA”和“低表达lncRNA-高表达miRNA-低表达mRNA”这2种调控轴构建ceRNA调控网络。
1.7 实时定量聚合酶链反应(qRT-PCR)分析
采用qRT-PCR分析lncRNA、miRNA和mRNA的相对表达水平,利用Trizol法[20]提取大麦Barke干种子和萌发24h的RNA样本,用大麦的actin基因作为lncRNA和mRNA的内参基因,以U6作为miRNA的内参基因,使用茎环法对miRNA进行反转录,引物序列见表1。

表1 荧光定量PCR引物信息Table 1 Primers for fluorescent quantitative PCR
2 结果与分析
2.1 lncRNA鉴定结果
对3个参试品种种子萌发的RNA-seq数据进行统计分析,发现46个文库总测序序列数量为11.4×108,质控处理后有效序列总数为11.2×108,其中有992426882个读数能比对到大麦参考基因组上。
为了识别大麦种子萌发的lncRNA,按照图1所示流程,使用Stringtie软件进行转录本重组,通过剔除已知的编码基因,获得了35245条候选lncRNA转录本,最后通过区分lncRNA和蛋白编码转录本,获得15053条可靠的lncRNA转录本。
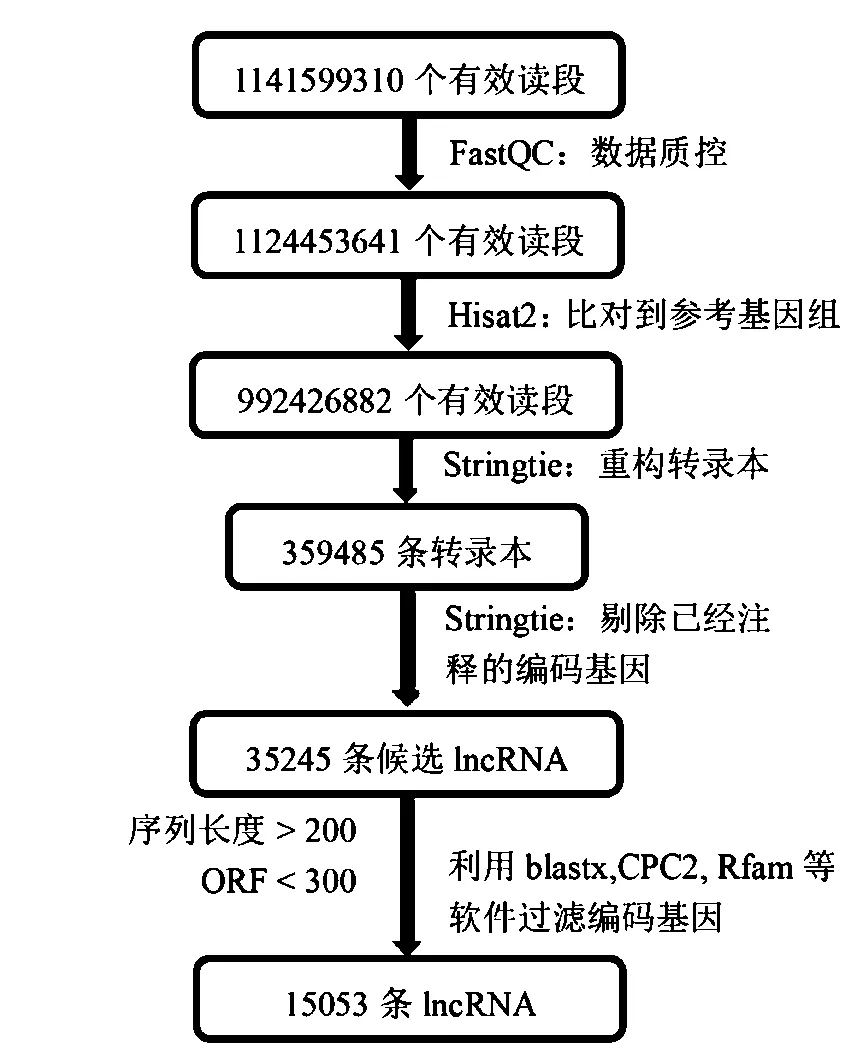
图1 大麦种子萌发 lncRNA鉴定流程 Fig.1 Identification processes of lncRNA inbarley seed germination
2.2 差异表达基因筛选
根据设定的筛选条件(P<0.05;|log2Foldchange|>2),从PRJNA577343供试大麦品种中筛选到17369个差异表达的mRNA和3523个差异表达的lncRNA,从PRJNA578897的品种IK621筛选到8976个差异表达的mRNA和1317个差异表达的lncRNA,品种IK573筛选到10307个差异表达的mRNA和1172个差异表达的lncRNA,其中3个品种共同的差异表达mRNA有6447个,共同的差异表达lncRNA有661个(见图2)。同时,通过miRNA Count数据,鉴定到310个差异表达的miRNA。

图2 不同品种大麦种子萌发差异表达的mRNA和lncRNA比较Fig.2 Comparison of differentially expressed mRNA and lncRNA in seed germination of different barley varieties
2.3 加权基因共表达网络构建及特异性模块鉴定
利用WGCNA对3个品种共有的差异表达的mRNA和lncRNA分别进行共表达分析,基于模块与种子萌发的相关性(R2>0.6)筛选特异性共表达模块。
在差异表达的mRNA分析中,设置权重值β为24,结果如图3所示。不同模块用不同颜色表示,共获得10个模块(Grey 模块表示未分配到任何模块的基因)。其中,brown模块包含的基因个数最多,有2246个;grey模块包含的基因个数最少,有8个,通过共表达网络与种子萌发进行关联,鉴定到与种子萌发极显著正相关的模块greenyellow(R2=0.68)(见图4),该模块包括1433个mRNA。
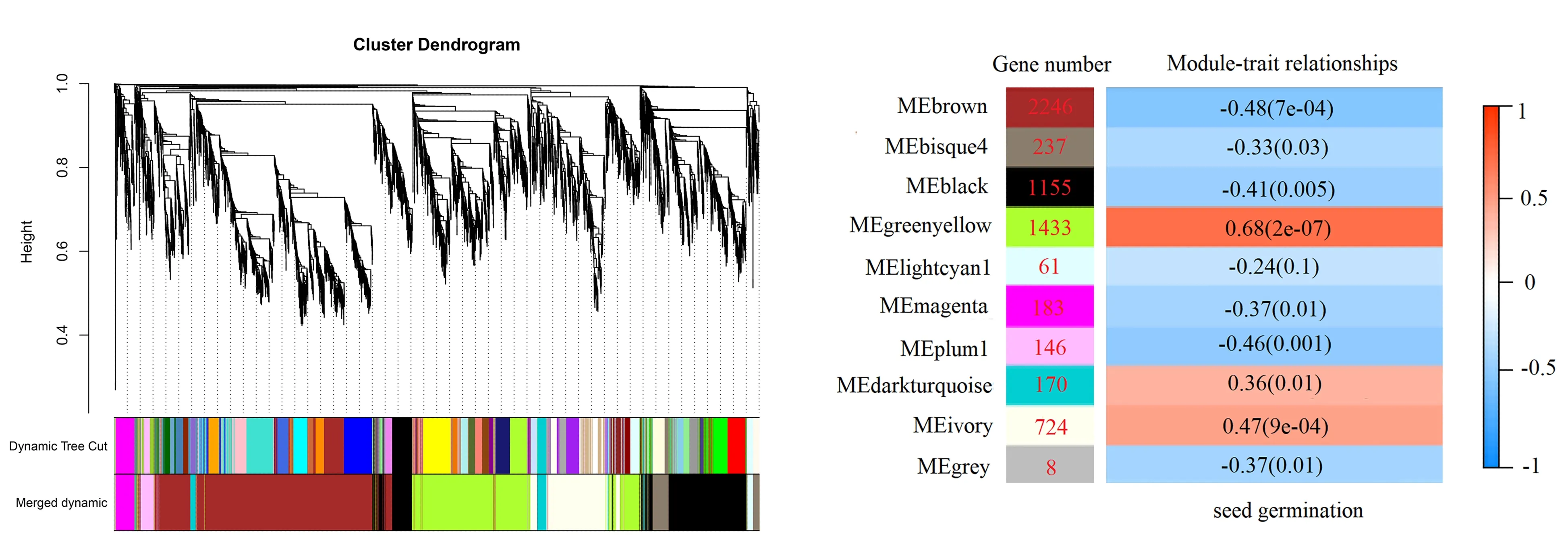
图3 mRNA聚类树及模块构建 图4 mRNA模块及其与种子萌发相关性 Fig.3 mRNA clustering tree and module construction Fig.4 mRNA module and its correlation with seed germination
在差异表达的lncRNA分析中,设置权重值β为12,结果如图5所示。通过lncRNA获得6个模块。其中,blue模块包含的基因个数最多,有325个;grey模块包含的基因个数最少,有10个,通过共表达网络与种子萌发进行关联分析,鉴定到与种子萌发极显著正相关的模块brown(R2=0.61)(见图6),该模块含有116个lncRNA。
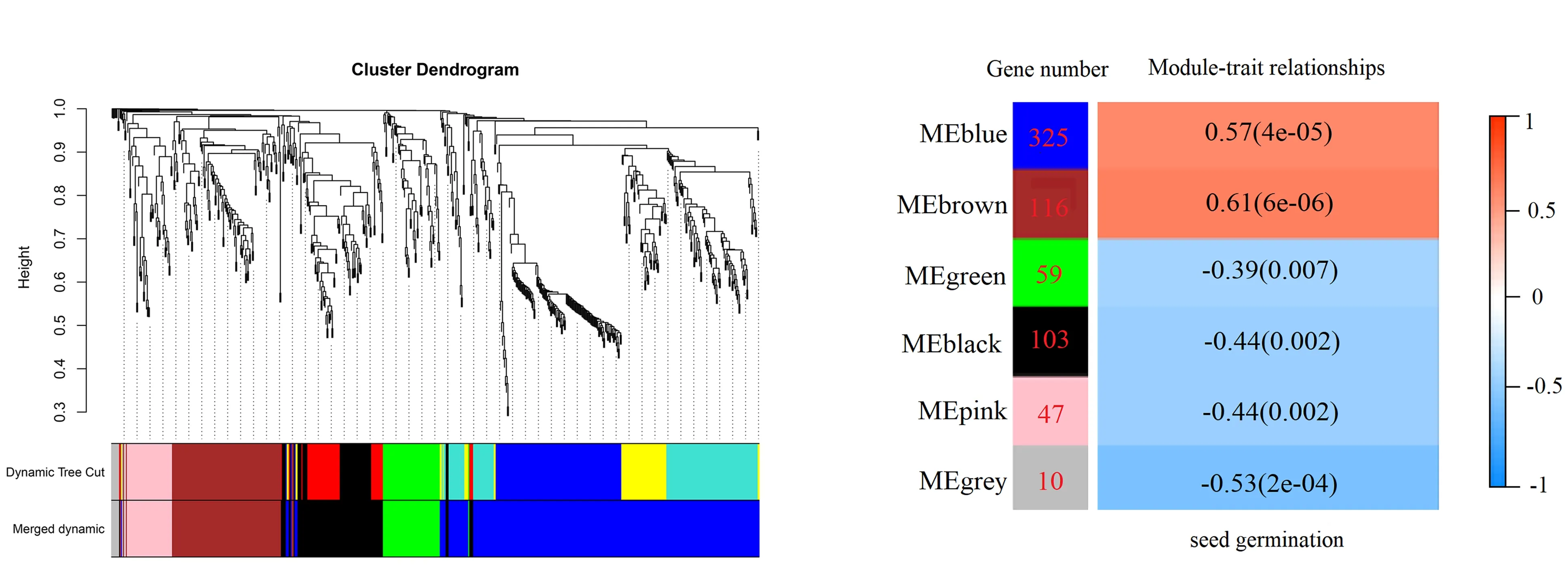
图5 lncRNA聚类树及模块构建 图6 lncRNA模块及其与种子萌发相关性 Fig.5 lncRNA clustering tree and module construction Fig.6 lncRNA module and its correlation with seed germination
2.4 mRNA特异性模块富集分析和通路分析
为进一步解析greenyellow模块的生物学功能,利用agriGO工具对模块内所有基因进行GO富集分析,这些GO通路(见图7(a))涉及到生物学过程、分子功能以及细胞组分。分析结果表明,该模块共富集到26个生物过程,其主要涉及水分响应(GO:0009415)、对酸性化学物质的响应(GO:0001101)、对无机物的响应(GO:0010035)、对含氧化合物的反应(GO:1901700)、胚发育(GO:0009790)等通路以及一些酶活通路,如肽酶调节活性(GO:0030414)、肽酶抑制剂活性(GO:0004866)、内肽酶调节活性(GO:0061135)、内肽酶抑制剂活性(GO:0061134)等。KEGG富集分析表明,greenyellow模块共有67个基因富集到7条通路,主要涉及MAPK信号转导通路、植物激素信号转导通路等(见图7(b))。
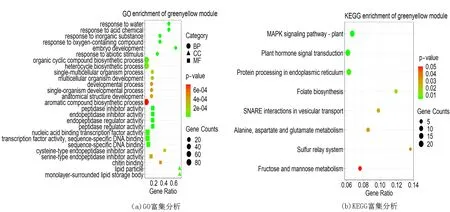
图7 greenyellow模块的GO和KEGG富集分析Fig.7 GO and KEGG enrichment analysis of greenyellow module
2.5 ceRNA网络构建结果
利用上述筛选到的mRNA、lncRNA与miRNA进行靶基因预测,获得168个miRNA-mRNA靶基因对以及310个miRNA-lncRNA靶基因对,最终根据“高表达lncRNA-低表达miRNA-高表达mRNA”和“低表达lncRNA-高表达miRNA-低表达mRNA”这2种调控轴构建ceRNA调控网络(见图8),该网络包含38个lncRNA、16个miRNA以及18个mRNA。其中“高表达lncRNA-低表达miRNA-高表达mRNA”的子网络包含了4个lncRNA、2个miRNA以及1个mRNA,显示基因HORVU0Hr1G039470的表达可能受到2个miRNA和4个lncRNA调控。“低表达lncRNA-高表达miRNA-低表达mRNA”子网络包含34个lncRNA、14个miRNA以及17个mRNA,其中miR854的表达受到19个靶向基因的调控,包括3个mRNA和16个lncRNA;而miR5054、miR5059、miR5060、miR5304、miR6432以及miR916的靶向lncRNA和mRNA均只有1个。此外,对ceRNA调控网络中的mRNA在大麦中进行了注释和在拟南芥中进行了同源基因的注释(见表2)。

表2 ceRNA调控网络中mRNA功能注释Table 2 mRNA function annotations in the ceRNA regulatory network
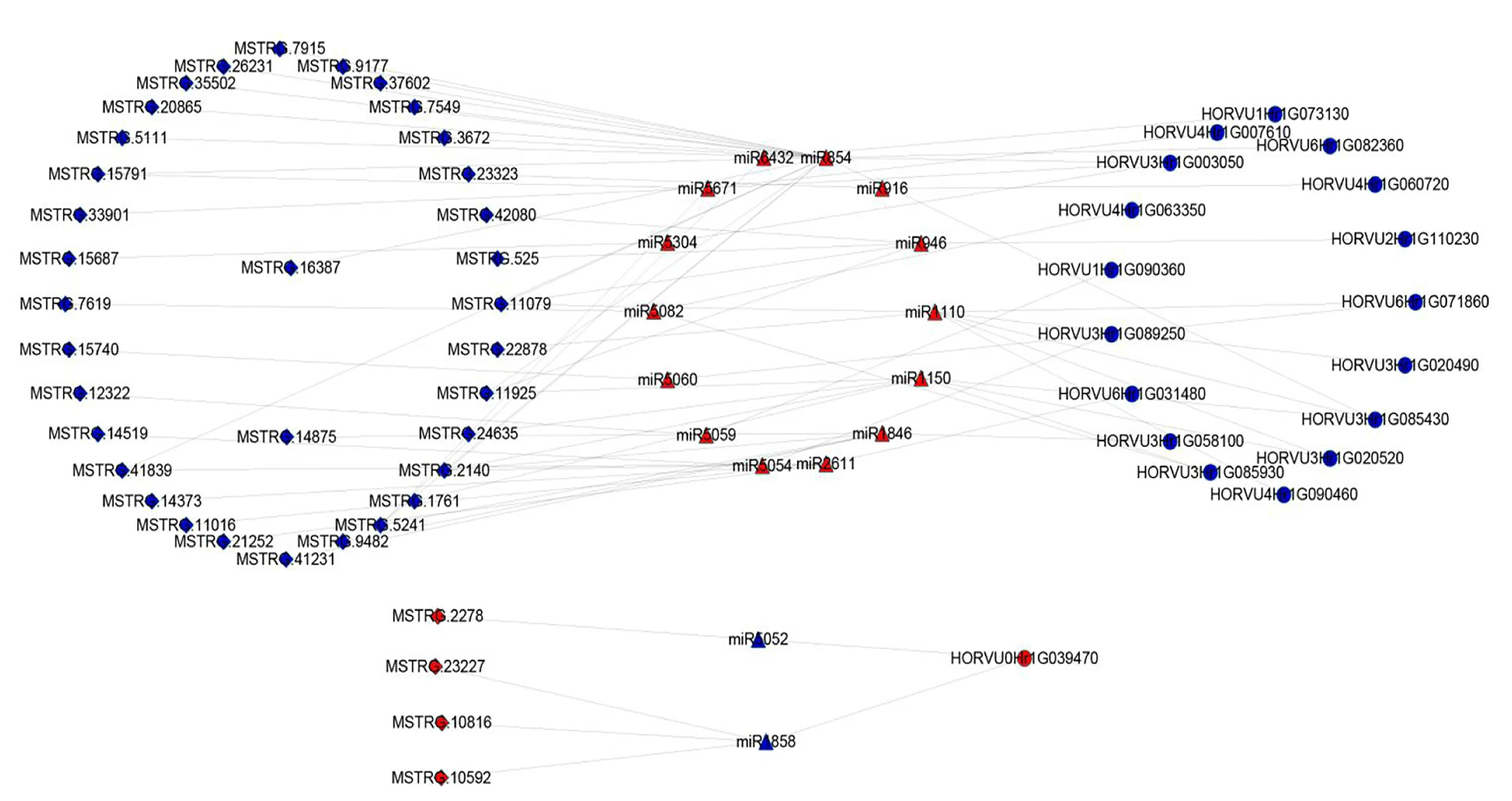
图8 大麦种子萌发ceRNA网络调控图Fig.8 ceRNA network regulatory map of barley seed germination
2.6 qRT-PCR分析结果
利用qRT-PCR检测基因HORVU0Hr1G039470、HORVU6Hr1G031480、MSTRG.23227、MSTRG.21252、miR1858以及miR2611的相对表达水平(见图9)。结果显示,HORVU0Hr1G039470、MSTRG.21252以及miR1858上调,基因HORVU6Hr1G031480、MSTRG.23227以及miR2611下调,这些基因的上下调模式与预测的ceRNA调控网络中的上下调模式一致。由此,初步验证了ceRNA调控网络的可行性,同时初步验证了HORVU0Hr1G039470-miR2611-MSTRG.21252和HORVU6Hr1G031480-miR1858-MSTRG.23227这2个关系对具有相应的ceRNA功能。
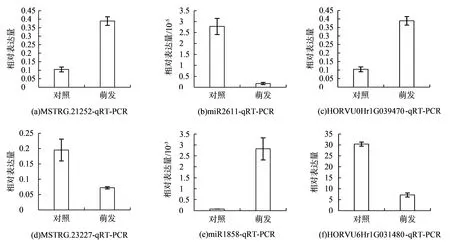
图9 大麦种子萌发过程中不同RNA的表达水平Fig.9 Expression levels of different RNAs during barley seed germination
3 讨论与结论
种子萌发是一个复杂的生物学过程,对植物的生存和繁衍至关重要。本研究通过对大麦萌发转录组数据进行分析,鉴定到了15053个lncRNA。通过差异表达基因分析,得到6447个差异表达的mRNA以及661个差异表达的lncRNA以及差异表达310个miRNA。对差异表达的mRNA和lncRNA进行WGCNA分析,分别鉴定到1个与大麦种子萌发高度相关的共表达模块,在这2个模块中,分别包含了1433个mRNA和116个lncRNA。高度相关的共表达模块中有67个mRNA被富集到MAPK信号通路,植物激素信号转导通路等KEGG通路上,这些通路均已证明与种子萌发有关[4]。通过对筛选到的基因进行靶基因预测,成功构建了一个包含2个子网络的ceRNA网络图,包含16个miRNA节点,38个lncRNA节点以及18个mRNA节点。
在“高表达lncRNA-低表达miRNA-高表达mRNA”子网络中,作为糖基转移酶基因HORVU0Hr1G039470的表达可能受到miR5052、miR1858以及4个lncRNA的调控。前人研究表明,糖基转移酶与种子的萌发有关[21],miR1858与籽粒发育有关[22],籽粒良好的发育是种子萌发的关键。MSTRG.2322或MSTRG.10816或MSTRG.10592可能通过竞争性地结合miR1858来调节HORVU0Hr1G039470的表达,从而调节大麦种子萌发过程。在“低表达lncRNA-高表达miRNA-低表达mRNA”子网络中,miR5054、miR5671及miR854与调控MAPK信号通路或植物激素信号转导通路相关[23-25],miR946参与种子的萌发以及休眠[26]。MSTRG.14519与HORVU3Hr1G089250可能竞争性地结合miR5054,从而影响种子萌发过程中激素的表达;MSTRG.33901或MSTRG.5241或MSTRG.15791能够与miR5671进行结合,从而调控谷氨酸代谢相关的基因HORVU4Hr1G007610、HORVU3Hr1G003050;与种子萌发相关的miR946以及HORVU2Hr1G110230通过与MSTRG.42080或MSTRG.11925或MSTRG.11079或MSTRG.525行使ceRNA功能,从而对大麦种子萌发产生影响。此外,miR854受到的靶向调控基因最多,说明该基因可能是大麦种子萌发过程中一个极为重要的节点miRNA。
本研究通过qRT-PCR验证了ceRNA调控网络中的2个关系对,其中HORVU0Hr1G039470和MSTRG.21252上调,而miR2611在大麦种子萌发中下调,基因HORVU6Hr1G031480、MSTRG.23227下调,而miR1858在大麦种子萌发中上调。这表明MSTRG.21252上调和miR2611下调可能促进miR2611对靶基因HORVU0Hr1G039470的促进作用,而MSTRG.23227下调和miR1858上调可能促进miR1858对靶基因HORVU6Hr1G031480的抑制作用。
综上所述,本研究鉴定出大麦种子萌发相关的差异表达基因,构建了大麦种子萌发相关的lncRNA-miRNA-mRNA互作ceRNA调控网络,初步揭示了3种RNA在大麦种子萌发中的调控模式。这些结果将有助于揭示种子萌发的分子调控机制,为种子萌发lncRNA、mRNA以及miRNA功能的进一步研究奠定基础。
猜你喜欢
作文小学高年级(2022年5期)2022-06-16
环境卫生工程(2021年4期)2021-10-13
四川轻化工大学学报(自然科学版)(2021年1期)2021-06-09
疯狂英语·新读写(2021年2期)2021-02-25
汉字汉语研究(2020年2期)2020-08-13
电子制作(2019年22期)2020-01-14
学苑创造·A版(2020年12期)2020-01-07
中国外汇(2019年15期)2019-10-14
疯狂英语·新读写(2018年3期)2018-11-29
作文教学研究(2016年1期)2016-07-05
