
呼伦贝尔草甸草原土壤古菌群落多样性高通量分析
2022-10-28 09:44:00高明华于春艳刘金月于凤莅孙广霁
饲料博览 2022年5期
高明华,于春艳,刘金月,于凤莅,孙广霁
(呼伦贝尔学院生命科学学院,内蒙古 呼伦贝尔 021008)
草地生态系统是由植被生态系统、土壤生态系统和微生物生态系统组成,其分布广泛,面积巨大,是重要的自然资源。内蒙古呼伦贝尔草原为温带草原,具有极其重要的生态服务功能、蕴含着巨大的碳汇资源,同时还是绿色畜产品的生产地和优质牧草的输出地。因此如何客观评价草地的利用与保护之平衡,就显得尤为重要。放牧和刈割是天然草场管理和利用的主要方式,也是两个主要人为干扰因素[1]。放牧和刈割一方面影响着植被生态系统,另一方面还会对土壤微生物产生的影响。近年很多天然草地实施了围封管理,研究表明,围封也是保护草地生态系统的重要措施之一。
微生物是草地生态系统的重要组成部分,推动了土壤的发生和发育,是生态系统物质与能量交换的重要纽带,维系着草地生态系统的可持续发展。土壤微生物学是生命科学与地球科学的新兴学科与交叉前沿,是地球元素生物化学循环的引擎[2]。同时,土壤微生物在草地生态系统的合理利用和土壤生态系统的保护方面也具有指示和指标作用。土壤微生物的多样性与群落结构是当今生物多样性研究的热点[3]。
古菌域是不同于细菌域与真核生物域的独立生命域,也是微生物的重要组成部分。土壤细菌和土壤真菌的群落结构、多样性以及在维持土壤肥力和可持续发展方面的研究已开展多年,但是古菌在土壤中的生物多样性及功能近些年才被关注。目前国内古菌群落研究可见于水生态系统[4]、农田土壤[5-6]等,关于草甸草原土壤古菌研究尚未有报道。本研究采用高通量测序技术(High-throughput sequencing,HTS),分析了不同利用方式下呼伦贝尔草甸草原土壤古菌群落的结构、多样性和功能,以揭示不同利用方式对草原影响的微生物学本质和古菌之作用,为草原土壤微生物多样性保护和利用提供理论依据。
1 研究区概况
试验样地位于呼伦贝尔草甸草原腹地,地理位置为E119°36′326″~119°59′856″,N49°17′781″~50°19′991″,海拔629~635 m。植被群落的主要物种有羊草(Lepidum chinense)、贝加尔针茅(Stipa baicalensis)等。试验地区属于中温带半干旱大陆性季风气候,年降水量356 mm,主要集中在7—9月份,变动幅度较大;年均气温-2.36℃,最高气温36.8℃,最低气温-48.1℃。年积温1 580~1 860℃,无霜期112 d。土壤类型是暗栗钙土。
2 研究方法
2.1 土壤样品的采集与处理
于2018年植物的生长旺季(8月份)在呼伦贝尔草原上进行野外观测及采样。试验样地(3块)位于呼伦贝尔草甸草原,利用方式分别为刈割(Y)、放牧(F)和围封(W),每块样地设置3个样方(20 m×20 m),每个样方内采用5点取样法采集0~10 cm土屋(分别为Y1、F1和W1)和10~20 cm(分别为Y2、F2和W2)土层样品混合成1个土壤样本,除去样本中的砾石和植物残根等,混匀、研碎,过2 mm筛,取部分土样用无菌塑封袋密封,置于0℃以下保温盒中带回实验室用于高通量测序分析。试验样地相关信息见表1。

表1 样本采样地相关信息
2.2 试剂与仪器
E.Z.N.A.TMMag-Bind Soil DNA Kit(型号为M5635-02),购于Omega Bio-Tek公司;分光光度仪(型号为NanoDrop-ND 1000),购于凯乐博(北京)科技发展有限公司;土壤DNA提取试剂盒购于Omega公司。
2.3 土壤基因组DNA提取及高通量测序
土壤碾碎过80目筛后提取土壤总DNA。利用土壤DNA提取试剂盒提取DNA,然后采用分光光度仪测定提取的DNA浓度,并利用1%的琼脂糖凝胶电泳检测DNA提取质量。将提取质量合格的DNA样品送生工生物工程(上海)股份有限公司对细菌16S rRNA V3~V4区进行测序,测序使用Illumina(MiSeq)平台。
2.4 生物信息学分析
原初数据经质控过滤,得到各样本有效数据。在97%的相似水平下利用QIIME(v1.8.0)软件进行物种操作分类单元(Operational taxonomic units,OTU)的划分,并进行基于OTU的物种组成分析、Alpha多样性分析、样本聚类和样本间相似性比较及基于COG(Clusters of orthologous groups,直系同源基因群)的功能预测分析等。
2.5 数据的统计与高通量分析
2.5.1 数据的统计
采用Excel 2011软件和SPSS 19软件进行数据的统计和分析。
2.5.2 高通量分析用软件和数据库
采用RDP数据库(Release 11.1,http://rdp.cme.msu.edu/)及R(v3.2)、Mothur 1.30.1和UniFrac软件等进行高通量分析。
3 结果与分析
3.1 测序结果质量分析
每个样品的有效序列数量测定结果见表2。
通过对6个不同处理的土壤样品进行Illumina MiSeq高通量测序,根据结果对序列进行统计,每个样品的有效序列数量结果见表2。原始序列的数目为481 319条,QC后得到优质序列的总数是468 904条,过滤后有效率为96.73%~99.25%。

表2 样品序列数统计结果
通常在97%的相似水平下对序列进行OTU的聚类,统计获得全部样品在不同OTUs中的丰度信息,共产生2 523个OTUs。6个土壤样品的优质序列长度主要分布在377~379 bp,在378 bp的序列最多为85 553条。
6个处理所含的古菌群落OTUs韦恩图见图1。由韦恩图可知,6个处理共有的OTU为65个,占比为13.18%~20.57%;而每个样品特有的OTU数量为23~41个,占比相对较低。W2特有的OTU在6个处理中最多,为41,占该样品总OTU的8.82%;Y1特有的OTU在6个样品中最少,为23,仅占该样品总OTU的6.37%。这说明不同处理方式对土壤古菌群落的影响很大;同时还表明,由于处理间与土层异质性增加,为种群的进化和演替提供了更大的可能。

图1 6个处理土壤古菌群落OTUs韦恩图
6个样品基于OTU的样本聚类树图见图2。样本聚类树图利用树枝结构能够更加直观地反映出多个样品之间的相似性和差异关系。树枝的长度代表样本间的距离,越相似的样本会越靠近。由图2可知,Y1与F1的相似性最高,W1与Y1、F1在一个分支上,说明在0~10 cm土层中放牧、刈割和围封处理方式下,土壤古菌群落均具有较高的相似性;Y2与F2的相似性次之,二者与W2不在一个分支上,W2与Y2、F2的相似性明显低于W1与Y1、F1的相似性。OTU的样本聚类树图还表明,W2与另外两个分枝(Y2和F2、Y1和F1)及W1分枝共同聚类在一起。总体分析来看,基本上反映出Y区与F区的古菌群落相似程度高,但与W区的相似程度较低。

图2 基于OTU的样本聚类树图
3.2 Alpha多样性分析
3.2.1 基于Alpha多样性的稀释曲线
W1、W2、F1、F2、Y1和Y2优质序列分别包含84 447、73 667、80 606、85 553、69 969、74 662条。使用97%相似度的OTU,利用Mothur软件进行Rarefaction分析,再利用R软件制作稀释曲线图[7]。各个样本稀释曲线见图3。
从图3中可以看出,序列数目达到400时各种样品稀释曲线均趋于平展,表明测序数据量合理,能真实反映土壤样本中的古菌群落。

图3 不同区组土壤古菌测序的稀释曲线
3.2.2 Alpha多样性指数
Alpha多样性指数见表3。

表3 6个土壤样品古菌多样性指数
由表3可知:各样本文库的覆盖率均为100%,说明土样中的基因序列能被检出的概率高,本次测序结果能代表草甸草原土壤古菌群落的实际情况。
根据香浓指数分析,古菌物种的多样性表现为W2>F2>F1>Y1>W1>Y2;在 刈 割 条 件 下,0~10 cm土层物种多样性则高于10~20 cm土层的。而在围封和放牧条件下,10~20 cm土层物种多样性均高于0~10 cm土层的;说明不同处理方式下土壤古菌演替存在不一致性。
基于土层方面,0~10 cm土层物种多样性表现为F1>Y1>W1;10~20 cm土层物种多样性表现为W2>F2>Y2。说明放牧和刈割在促进物种丰度和群落多样性增加方面具有积极的作用。
根据Chao1指数分析,各处理物种的丰度表现为W2>F1>W1>F2>Y1>Y2;在 刈 割 和 放 牧 条 件下,0~10 cm土层物种的丰度均高于10~20 cm土层的,而围封条件下,10~20 cm土层物种的丰度则高于0~10 cm土层的(W2>W1)。
基于土层方面,0~10 cm土层物种丰度表现为:放牧>围封>刈割;10~20 cm土层物种丰度表现为:围封>放牧>刈割。
通过表3的综合数据看来,W2区的古菌群落多样性和丰富度都最高,因此,围封对保持物种丰度和群落多样性具有重要作用。
从不同处理间Alpha指数箱式图可以看出,样本内的离散程度,例如从辛普森指数来看样本之间的离散程度结果见图4。
从图4可以看出,F区组的盒子最窄,说明该区内样本离散程度最小,相比较而言具有较高的保守性;W区组的盒子最宽,说明该区样本的离散程度最大,W区组的古菌群落相对于F区与Y区保守性较低,结果表明围封有利于古菌群落多样性的保护,而重度放牧时古菌群落多样性则明显降低。

图4 处理间Alpha指数箱式图(辛普森指数)
3.3 土壤样品古菌群组成分析
从门水平分析群落组成,结果见图5;从属水平分析群落组成,结果见图6。
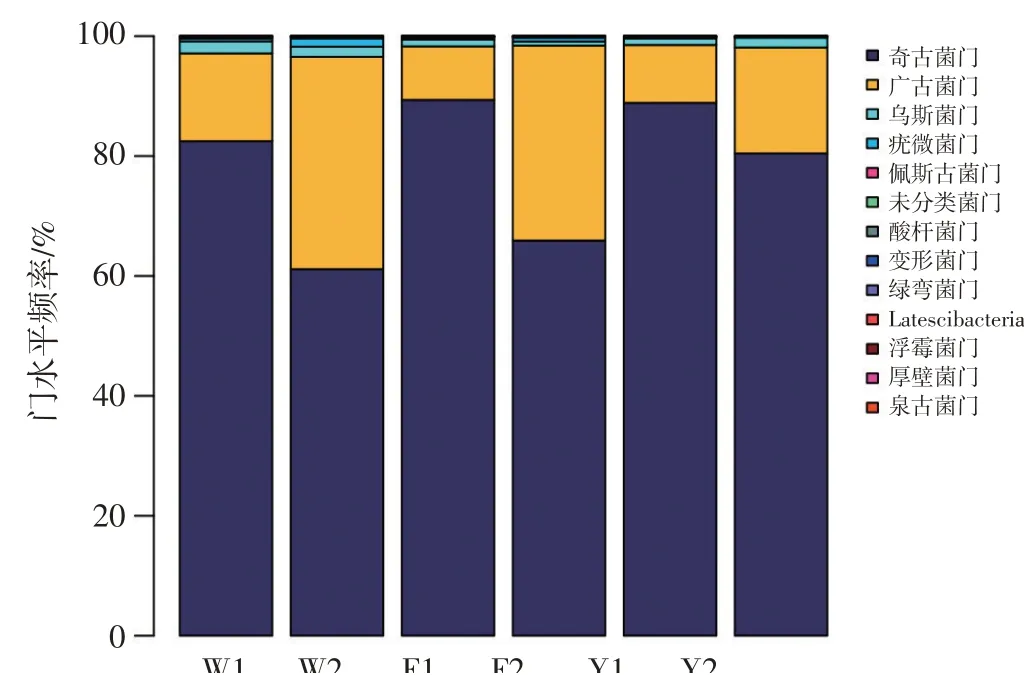
图5 门水平所有样本群落结构分布情况

图6 属水平所有样本群落结构分布图
从门的分类水平看,6个土壤样品中共检测出12个门类,其中奇古菌门(Thaumarchaeota)、广古菌门(Euryarchaeoya)平均占比分别为78.03%和19.81%,为优势菌门;乌斯菌门(Woesearchaeota)和疣微菌门(Verrucomicrobia)平均占比分别为1.37%和1.12%,是主要菌门,其他菌门占比均低于1%。
由图6可知,6个土壤样品中共检测到24个属,其中亚硝化球菌属(Nitrososphaera)、产甲烷类球菌属(Methanomassiliicoccus)平均占比分别为78.03%和19.39%,为优势属;乌斯菌AR16属(Woesearchaeota Incertae SedisAR16)、斯巴杆菌属(Spartobacteria genera incertae sedis)平均占比超过1%,是土壤古菌
如图8所示,除了W1与W1这样的自身交叉区的颜色之外,F1与Y1交叉区的颜色是最深红的,所以二者古菌群落组成最相似,其次是F2与W2、W1与Y2、W1与Y1、W1与F1、Y1与Y2交叉区的颜色逐渐变浅,说明其间的相似性依次变低;F1与Y2、W2与F2、W2与Y2、W1与W2、F2与F1、F1与F2交叉区的颜色由浅蓝依次变为深蓝,说明其间的差异性依次递增。W2与F1、Y1交叉区的颜色最为深蓝,所以W2区与F1区、Y1区古菌群落组成差异性最大,相似水平最低。故通过对Unifrac分析结果的聚类发现,F区与Y区的古菌群落相似性高。二者与W区组的古菌群落的相似性低。中的主要群落。
3.4 主成分分析
样本间的相似度越高则在图中的位置越聚集,基于OTU的主成分分析(Principal component analysis,PCA)三维图结果见图7。

图7 基于OTU的PCA三维图
如图7所示,可以看出PCA1、PCA2和PCA3的样品差异性贡献率分别达到93%、4%和2%,合计达到99%,是差异的主要来源,根据各样本在图中的位置关系可以看出,F1区与Y1区更加聚集些,所以F1区与Y1区的相似度高,其次为W1和F2。
3.5 样品间复杂度比较分析
各样本之间距离关系分析结果见图8。
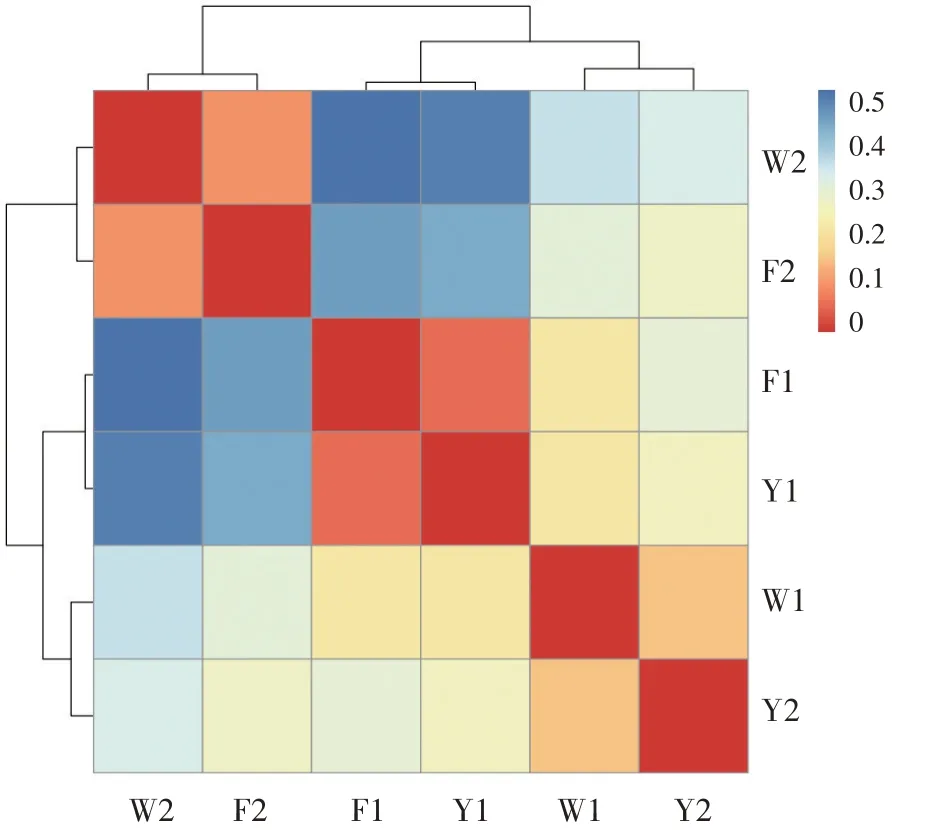
图8 样品距离热图(加权UniFrac的热图)
3.6 功能测序分析
样品中古菌的功能预测结果见图9。
根据功能测序结果,可观测各样品土壤古菌在更高层级水平上的功能。由图9可以看出,6个样品在更高层级上的功能主要有25种,其中能量产生与转换(Energy production and conversion),通用一般功能预测(General function prediction only),复制、重组与修复(Replication,recombination and repair),核苷酸的迁移和代谢(Migration and metabolism of nucleotides),翻译、核糖体结构和生物发生(Translation,ribosomal structure and biogenesis)以及氨基酸的运输和代谢(Transport and metabolism of amino acids)等功能在古菌群落中表现明显。同时由图9可知,三种处理0~10 cm土层土壤古菌的功能明显强于10~20 cm土层的,说明0~10 cm土层土壤古菌的代谢活性强于10~20 cm的。F1和Y1、W2和F2及Y2和W1在主要功能和功能强度上具有相似性,说明放牧和刈割对0~10 cm土层古菌的功能影响比较明显。
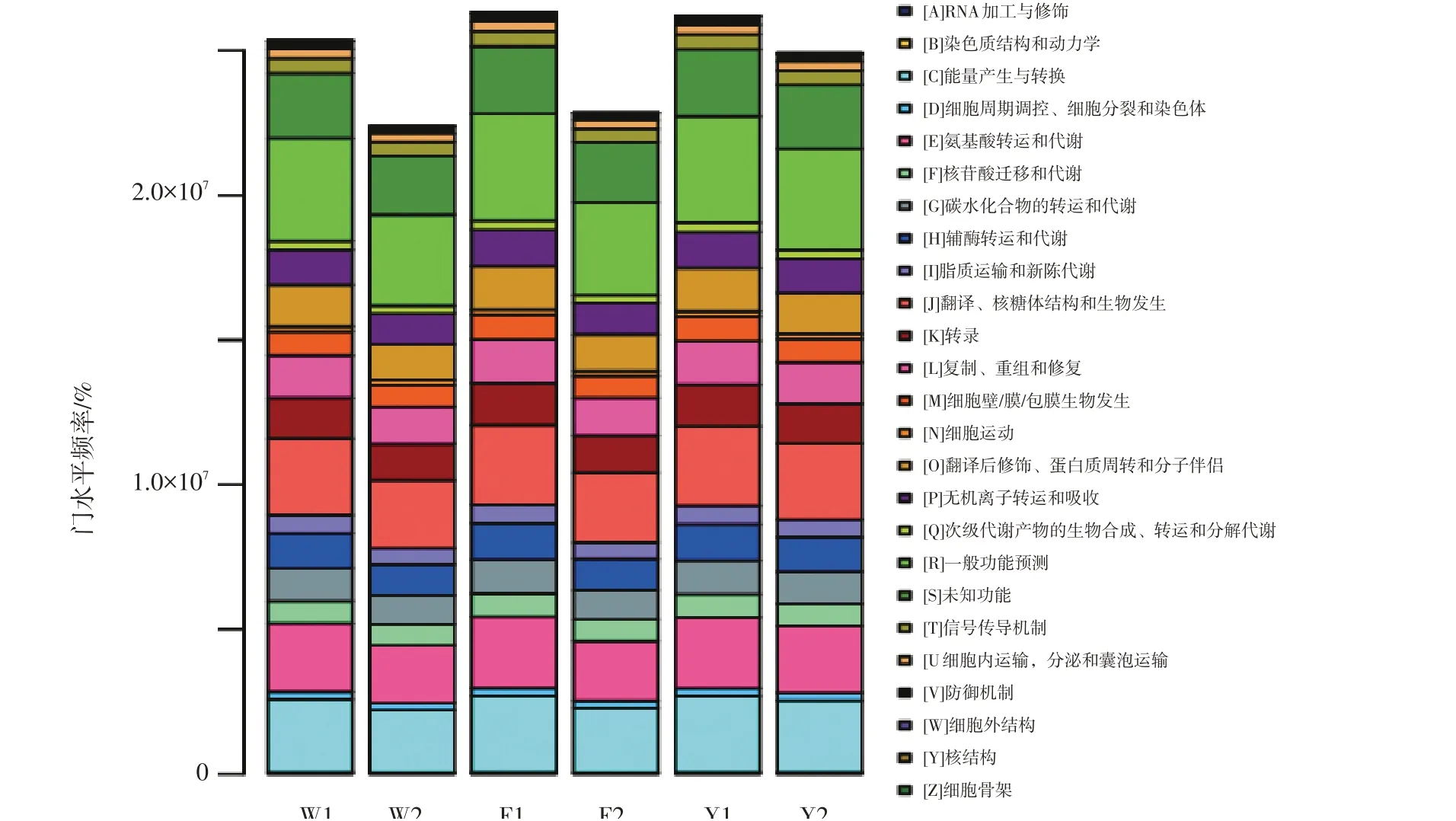
图9 基于COG通路的功能结构分布柱状图
4 讨论
一般来讲,古菌主要分布在极端环境,但在一些非极端环境中均有分布,其分布的生态类型多样,包括水生态系统[4]、农田土壤[5-6]、放射污染区[7]、重金属污染区[8]、火山喷发沉积物[9]和陆坡沉积物[10]。研究结果显示,目前已检测到包括奇古菌、广古菌和泉古菌等12个门21个科36个属。这说明古菌存在的生态环境类型多样,对各种环境条件具有很好的适应性,以保证各科属古菌群落的多样性和物种丰度,同时为研究草地生态系统古菌群落结构和物种多样性具有重要意义。本次试验土壤样本采集地为呼伦贝尔草甸草原腹地,气候特点为中温带半干旱大陆性季风气候,年降水量为356 mm,降雨主要集中在7—9月份,变动幅度较大;年平均气温-2.36℃,最高气温36.8℃,最低气温-48.1℃。年积温1 580~1 860℃,无霜期112 d。土壤类型是暗栗钙土。
土壤理化指标比较均衡[11],不同处理方式对古菌群落结构和物种多样性与丰度均有一定的影响。
土壤微生物群落功能多样性是反映土壤生态系统功能与稳定性的重要指示因子。随着新一代高通量测序技术的广泛应用,已成为土壤质量评价的常规方法。但对土壤古菌群落多样性研究的还相对较少,尤其是对草甸草原土壤中古菌群落特征分析尚未有报道,本研究通过对6个不同处理的研究区的土壤样品进行Illumina MiSeq高通量测序,根据结果对序列进行统计,得到优质序列468 904条。在97%的相似水平下进行OTU的聚类,以获得全部样品在不同OTUs中的丰度信息,共有2 523个OTUs,共有的OTU为65个,占比为13.18%~20.57%。研究区的古菌被分为13个门。但不同生境古菌的群落结构和多样性方面还存在很大的不同。闫慧贞等[4]利用16S rRNA基因扩增子测序技术,研究了梅山岛海域春季浮游古菌群落空间分布情况,结果表明该海域浮游古菌在原核群落中的相对丰度为0.6%~26.5%,浮游古菌群落由奇古菌门Marine GroupⅠ(MGⅠ)和广古菌门Marine GroupⅡ(MGⅡ)主导,MGⅠ的物种组成较为单一,而MGⅡ的系统发育多样性较高。张伟等[5]的研究结果表明,新疆干旱半干旱土壤环境下当地原生态土壤古菌群落主要由Thaumarchaeota、SM1K20、Euryarchaeota、Aenigmarchaeota、Crenarchaeota和Marine-HydrothermalVentGroup(MHVG)等6个门组成。虽然各样品特有OUT相差较大,但各样品共有的核心OTU数仍高达232个,占主要部分。古菌不仅从参与多种元素的地球化学循环中获得能量,而且成为数亿年地质形成过程中最重要的地质营力,所以地质生态环境的变化必定指示着古菌种群的多样性差异。
古菌和细菌和真菌一样,在土壤物质循环转化和生态系统平衡方面具有非常重要的作用。其作用是通过其各种功能来发挥的。目前在古菌的功能研究中,常用基于COG功能的预测分析,通过对宏基因组预测数据功能分析与对应16S预测功能分析结果的比较,表明该方法对土壤菌群功能分析的准确性接近89%以上,因此该方法能非常好地反映样本的功能基因构成[12]。本研究显示6个样本在更高层级上的功能主要有25种,涉及到能量代谢、核苷酸与核酸代谢、氨基酸和蛋白质代谢、核糖体结构与生物发生、细胞代谢等多方面,表现出其功能的多样性,这些多样性的功能与细菌和真菌的功能基本一致[13-18]。表明不同微生物的功能在分子层面上具有同质性。
5 结论
1)基于细菌群落多样性层面,6个样本表现为W2>F2>F1>Y1>W1>Y2;基于物种丰度层面,6个样本表现为W2>F1>W1>F2>Y1>Y2。
2)基于门和属层面,6个样本的优势菌门、优势菌属和其他菌门、菌属构成相似,但相对丰度存在一定的差异。
3)草原土壤古菌主要功能与细菌、真菌基本一致,说明基于分子层面不同微生物的功能具有同质性。
4)个样本土壤古菌群落结构与功能相似,具有一定的稳定性。植被类型、处理方式和土层深度等都是影响古菌群落结构和多样性的因素。
猜你喜欢
土壤学报(2022年3期)2022-08-26 12:15:26
建材发展导向(2022年10期)2022-07-28 03:04:22
大自然探索(2022年5期)2022-07-11 03:10:33
知识就是力量(2022年6期)2022-06-16 20:19:36
昆明医科大学学报(2022年2期)2022-03-29 00:51:58
食品安全导刊(2021年20期)2021-08-30 06:40:50
当代陕西(2020年24期)2020-02-01 07:06:40
当代陕西(2020年24期)2020-02-01 07:06:36
当代陕西(2020年24期)2020-02-01 07:06:36
水生生物学报(2015年1期)2015-02-28 16:01:05
