
Comprehensive bioinformatic analysis of key genes and signaling pathways in glioma
2022-10-24 01:50XiaomingZhangMengyuanJiangShenfengTangChaoshiNiuandShanshanHu
中国科学技术大学学报 2022年9期
Xiaoming Zhang ,Mengyuan Jiang ,Shenfeng Tang ,Chaoshi Niu ,and Shanshan Hu
1Department of Neurosurgery,the First Affiliated Hospital of USTC,Division of Life Sciences and Medicine,University of Science and Technology of China,Hefei 230001,China;
2Anhui Key Laboratory of Brain Function and Diseases,Hefei 230001,China
Abstract: The identification of specific survival-related differentially expressed genes (DEGs) is a method for uncovering therapeutic approaches for various cancers,including glioma.However,the key target genes associated with the occurrence and development of gliomas remain unknown.In this study,we performed bioinformatics analysis on 17 GSE datasets and identified DEGs correlated with glioma.A total of 74 mutual-DEGs with downregulated expression in gliomas compared with that in normal brain tissues were found in 17 datasets.These DEGs were related to GABAergic synaptic transmission,chloride transmembrane transport,glutamate secretion,and gamma-aminobutyric acid signaling pathway.Gamma-aminobutyric acid type A receptor subunit gamma 2 (GABRG2) was identified as a hub gene in the protein-protein interaction network.GABRG2 exhibited lower expression in IDH wild-type astrocytoma than that in IDH mutant astrocytoma and indicated poor prognosis in glioma patients.GABRG2 may contribute to the progression of glioma by affecting GABA receptor-related pathways and is a potential biomarker for the diagnosis and treatment of glioma.
Keywords: glioma;bioinformatic analysis;GABRG2;biomarker
1 Introduction
Glioma is a heterogeneously primary and malignant tumor of the central nervous system that contains multiple subtypes with different clinical and biological characteristics[1].According to the current World Health Organization (WHO) classification of central nervous system tumors,tumors can be divided into grades I-IV.Glioblastoma (grade IV) has the highest degree of malignancy and incidence[2].Despite having received maximal surgical treatment,postoperative radiotherapy,and chemotherapy,the prognosis of glioma patients remains poor,with a median overall survival time of only 12-15 months[3].Thus,glioma remains a serious threat to human health,and there is an urgent need for an in-depth study of the molecular mechanism of its occurrence and development.
With the rapid development of transcriptome sequencing and bioinformatics,RNA sequencing (RNA-seq) has also attracted widespread attention[4-6].Recent studies have shown that RNA-seq can be used not only to reveal the therapeutic targets of drugs but also to uncover the differential gene profiles in the progression of various diseases[4-6].However,largescale RNA-seq analysis of the pathogenesis of gliomas requires further investigation.
In the present study,we used bioinformatics to analyze differentially expressed genes (DEGs) in 948 samples of glioma tissue and 81 samples of normal brain tissue,aiming to discover DEGs correlated with the occurrence and development of glioma.We then performed gene ontology (GO) annotation,Kyoto Encyclopedia of Genes and Genomes (KEGG)analysis,and created a protein-protein interaction (PPI) network to analyze the DEGs.Lastly,we selected the key gene GABRG2 associated with the biological progress of glioma and explored the expression and clinical significance of GABRG2 in different subtypes of glioma.The results of this study aid the identification of a potential target for the clinical diagnosis and treatment of glioma.
2 Materials and methods
2.1 Screening the gene set enrichment (GSE) dataset
We searched for the keyword “glioma or glioblastoma,expression profile of array” in the Gene Expression Omnibus(GEO) database①https://www.ncbi.nlm.nih.gov/geoof the National Center for Biotechnology Information (NCBI)[7]and then selected “Homo sapiens”.The GSE datasets used in this experiment met the following criteria:(i) tumor samples collected from patients with glioma or normal brain tissue,(ii) prospective research,(iii) application to GPL570 platform,and (iv) raw data in CEL format.A flowchart depicting the data preparation,processing,and analytical processes is shown in Fig.1.
2.2 Basic information of the GSE datasets

Fig.1.Flowchart of data processing and DEG analysis.
Seventeen datasets (GSE108474,GSE28238,GSE43378,GSE73066,GSE61374,GSE53733,GSE50161,GSE44971,GSE45921,GSE36245,GSE33331,GSE26576,GSE15824,GSE19728,GSE9171,GSE5675,and GSE4290) were downloaded from the GEO database of NCBI[8-24].Then,we selected these datasets with clear pathological diagnosis samples for further study.A total of 81 normal brain tissue samples and 948 glioma samples (including 204 cases of WHO grade I,134 cases of WHO grade II,135 cases of WHO grade III,and 475 cases of WHO grade IV) were included in the study;663 cases were adult gliomas,and the remaining 285 cases were childhood gliomas.
2.3 Assessment of data quality and identification of DEGs
GEO2R①https://www.ncbi.nlm.nih.gov/geo/geo2r/[25]is an interactive web tool in the GEO database that uses the GEOquery and limma R packages of the Bioconductor project to compare the processed data;the DEGs between different condition groups can then be screened quickly and efficiently.Recently,GEO2R has been widely used in differential expression analysis of GSE datasets[26].
In this study,we used GEO2R to test the standardization of data in the 17 datasets,draw box plots,and clarify the data distribution in each sample.R software (version 3.5.2) was used to analyze the changed logFC of the DEGs between glioma and normal brain tissue in each dataset.The Benjamini and Hochberg method was used to calculate the adjustedPvalue of the differential genes (adj.P).We set |logFC| >2 and adj.P< 0.01 as the screening criteria to identify the DEGs.
2.4 Volcano map,Venn diagram,and heat map
The expression profiles of glioma DEGs were obtained using the R software (version 3.5.2).Graphpad Prism software (version 8.0) was used to create the volcano plots,and Bioinformatics Evolutionary Genomics was performed to cross-analyze the DEGs profiles of 17 GSE datasets and draw a Venn diagram to obtain the mutual-DEGs.In addition,the online tool“Morpheus” was used to draw a heat map of mutual-DEGs in gliomas.
2.5 Analysis of GO and KEGG pathway
GO analysis includes biological process (GO-BP),molecular function (GO-MF),and cellular component (GO-CC)[27].The KEGG database①https://www.genome.jp/kegg/has been widely used for the systematic analysis of gene functions[28].
The Database for Annotation,Visualization,and Integrated Discovery (DAVID)②https://david.ncifcrf.gov/is an online analysis tool used for the integrated analysis of multiple genes[29].We used DAVID(version 6.8) to perform GO analysis of DEGs and R software to draw the pathway diagrams.KOBAS3.0③http://kobas.cbi.pku.edu.cn/kobas3[30]was selected to analyze the DEGs (the critical threshold was set as the number of enriched genes > 2,P< 0.01).The first 10 enrichment pathways were selected as the final results,and a bubble chart was drawn using R software.
2.6 Construction of PPI network and selection of core modules
Glioma-related DEGs were uploaded to the web tool “Search Tool for the Retrieval of Interacting Genes” (STRING,version 11.0)④https://string-db.org[31]to obtain the interaction information between DEGs.We changed the species to “Homo sapiens” and set the confidence level > 0.4 to construct the PPI network.
Cytoscape software[32]was applied to analyze the PPI network,and the MCODE plug-in was selected to screen the key modules in the PPI network.The cytoHubba plug-in was used to calculate the basic information of each gene in the PPI network[33].Furthermore,the software Metascape⑤http://metascape.org/gp[34]was used to identify key modules and clarify their functions.P< 0.05 was considered as statistically significant.
2.7 Identification,survival,and expression analysis of hub genes
Using the “cytoHubba” plug-in in Cytoscape software to calculate the score of each gene in the core module according to the maximal-clique centrality (MCC) algorithm,10 genes with the highest scores were selected as the key genes.Next,the Gene Expression Profiling Interactive Analysis (GEPIA2)online web tool⑥http://gepia2.cancer-pku.cn[35]was used to analyze the impact of these 10 key genes on the prognosis of glioma patients.
We analyzed the expression of key DEGs in 81 normal tissues and 948 glioma tissues in 17 datasets using the Cancer Genome Atlas (TCGA) and Chinese Glioma Genome Atlas(CGGA)⑦http://www.cgga.org.cn[36]further clarified the expression trend of key DEGs associated with glioma and their expression in different molecular subtypes of glioma.Lastly,the GeneMANIA online web tool⑧http://genemania.org[37]was used to analyze the related pathways in which key genes may be involved in glioma.
2.8 Patients and clinical samples
The cohort of clinical specimens included 20 samples of glioma tissue and 15 samples of normal brain tissue obtained from the Department of Neurosurgery of the First Affiliated Hospital of USTC between February 2020 and October 2021.All tumor samples were pathologically confirmed to be glioblastoma.Normal brain tissues were obtained from patients who underwent brain tissue resection for acute craniocerebral injury.All specimens were immediately stored in liquid nitrogen.Besides,written informed consent was obtained from all patients,and the study was approved by the Ethics Committee of the First Affiliated Hospital of USTC.
2.9 Cell culture
Fresh glioblastoma specimens were collected from the operating room and immediately dissociated into single cells using 0.1% trypsin to obtain primary glioblastoma cells (named GP-1 and GP-2).Human glioma cell lines (U251,U87,T98G,TJ905,and LN229) and normal cells (HEB) were maintained in our laboratory.All cell lines were received mycoplasma contamination test and determined to be mycoplasma-free.All cells were cultured in high-glucose Dulbecco’s modified Eagle’s medium (DMEM,Hyclone) containing 10% fetal bovine serum (Clark) and stored in an incubator at a constant temperature of 37 °C at 5% CO2.
2.10 RNA extraction and real-time quantitative PCR(qRT-PCR)
Total RNA from glioma tissues and cells was extracted using TRIzol reagent (Invitrogen),according to the manufacturer’s protocols.Next,1 μg of RNA,quantified by NanoDrop ND-3300 (Thermo Fisher Scientific),was reverse-transcribed using the GoScript Reverse Transcription System (Promega)with the corresponding primers.Finally,qRT-PCR analyses were performed with TransStart Top Green qPCR SuperMix(+Dye II) (TransGen) on a LightCycler96 (Roche),and GAPDH was used as an internal control.
2.11 Statistical analysis
Statistical analysis was performed using GraphPad Prism software (version 8.0),and the results are expressed as mean ±standard deviation.The difference between the two groups of samples was calculated using the independentt-test,and the difference between the three groups of samples was determined using the chi-square test.P< 0.05 was considered to be statistically significant.
3 Results
3.1 Analysis of DEGs correlated with glioma in GSE datasets

Fig.2.Identification of 74 mutual differentially expressed genes (DEGs) in 17 GSE datasets.(a) Volcano plot of glioma-related DEGs in datasets.(b)Venn diagrams of glioma-related DEGs in four groups.(c) Venn diagram of mutual DEGs in 17 datasets.
We used the online tool GEO2R to analyze 17 GSE datasets and created a box plot to clarify the distribution of the samples in each dataset.The results showed that the distribution in the 17 datasets was centered on the median value and was essentially at the same level,suggesting that the samples included in this study had a high degree of standardization and the data were comparable (Fig.S1).Subsequently,the R software program was used to analyze the DEGs between the glioma tissue samples and normal brain tissues in each dataset with the threshold (adj.P< 0.01 and |logFC| > 2) (Figs.1,2a,and S2).Crossover analysis of these DEGs showed 74 overlapped genes (Fig.2b and c),namely SYNPR,RALYL,TMEM130,KCNJ6,FSTL5,PTPRR,C1QTNF4,SYT1,STYK1,GRM3,SMIM17,RBFOX3,SPOCK3,NEUROD6,ELAVL2,CYP4X1,KIAA0319,KIAA1107,GABRG2,GABRB2,PCP4,PAK7,FAM19A1,CA11,SYININGR3,FAM19A1,CA11,SYININGR3,GJNG6,NEAP,CDHERP,NEAP,GJNG3,NPY1R,TAC1,SST,C11orf87,WIF1,ANO3,CALB2,DNM1,GABRA2,CALY,RBFOX1,HTR2A,LINGO2,ERC2,HPCA,TRHDE,C3orf80,CPLX1,CACNG3,CCK,OLX1,CACNA RELN,NECA,GRIN,LOC285147,STMN2,VSNL1,VIP,SLC26A4-AS1,LINC00889,RYR2,KRT222,MAL2,PDE1A,GABRA1,UBE2QL1,SERTM1,NWD2,RSPO2,SLC12A5,and SCN3B.These DEGs may play important roles in the occurrence and development of glioma.In addition,we utilized the online tool Morpheus to process the 74 DEGs in 17 datasets and generated heat maps.The results showed that these 74 DEGs were significantly downregulated in glioma tissues(Figs.2 and 3).
3.2 Results of GO and KEGG analysis in mutual DEGs
To explore the possible action pathways of the common DEGs in glioma,we used the DAVID (version 6.8) to perform the GO analysis.The results showed that the 74 DEGs enriched the following processes:24 GO_BP processes,including chemical synaptic transmission,GABAergic synaptic transmission,and γ-aminobutyric acid signaling pathway (Fig.4a);19 GO_CC processes,including cell connection,axon,and GABA-A receptor complex (Fig.4b);and eight GO_MF processes,such as GABA-A receptor activity,extracellular ligand-gated ion channel activity,and GABA-gated chloride channel activity (Fig.4c).Subsequently,we used KOBAS(version 3.0) to perform KEGG analysis of common DEGs.The results showed that these common DEGs interacted with neuroactive ligand receptors,GABAergic synapses,and glutamatergic synapses (Fig.4d).
3.3 PPI network of mutual DEGs,core module filtering,and function enrichment
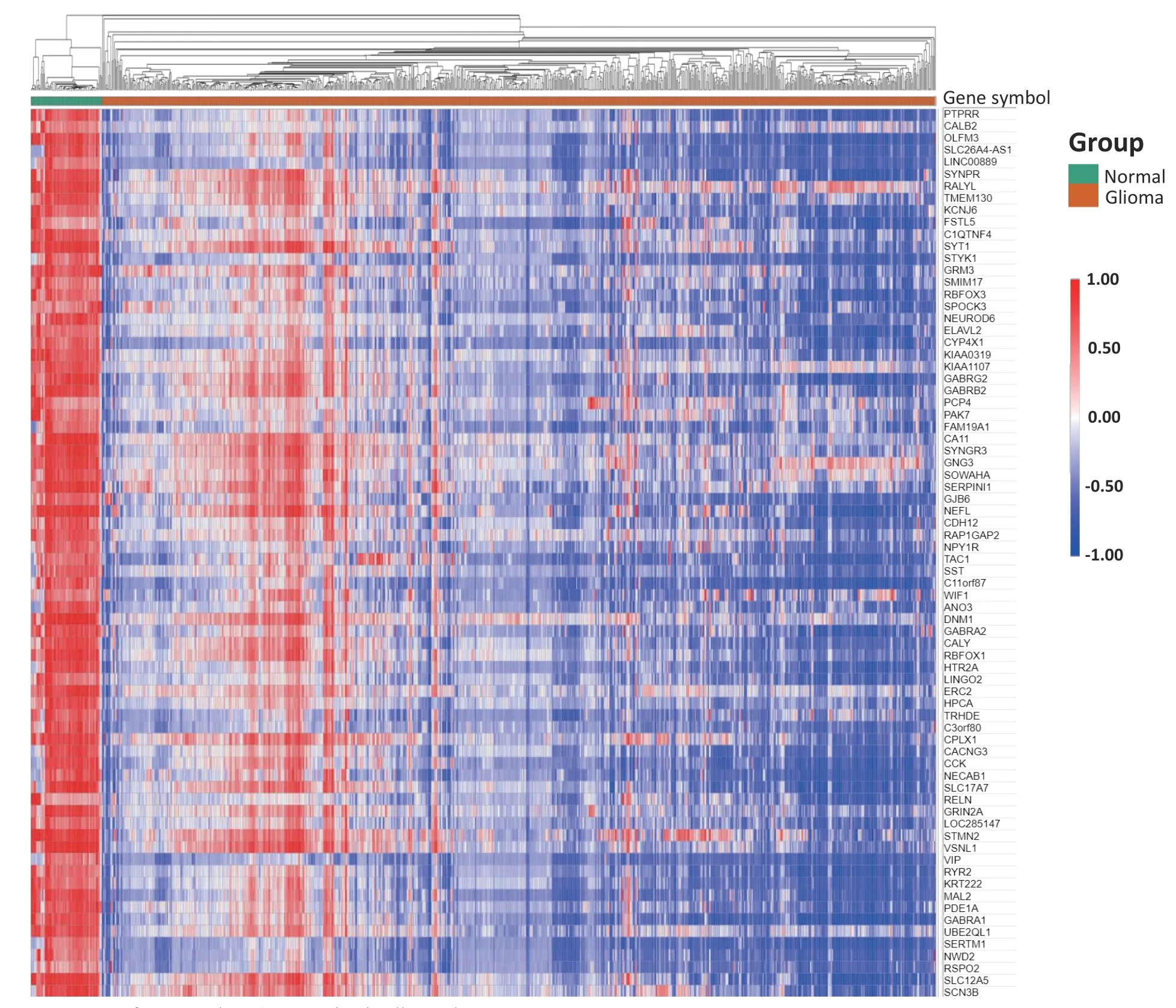
Fig.3.Heat map of 74 mutual DEG expression in all samples.

Fig.4.Functional enrichment analyses of mutual DEGs in 17 datasets.(a) Gene ontology (GO) biological process analysis of 74 mutual DEGs.(b) GO cellular component analysis of 74 mutual DEGs.(c) GO molecular function of 74 mutual DEGs.(d) Bubble chart shows the result of KEGG pathway analysis of 74 mutual DEGs.P < 0.05 was considered as statistically significant.
To investigate the interaction between the DEGs,we used STRING to construct a PPI network of the 74 common DEGs.We uploaded the PPI network to Cytoscape and used the cytoHubba plug-in to calculate the basic information of each node in the PPI network,describe the node based on the obtained degree and betweenness centrality,and apply it to the combined score to determine the interaction between each node (Fig.5a).Subsequently,we used the MCODE plug-in in Cytoscape to analyze and screen the key modules in the PPI network and found a module composed of 17 genes(Fig.5b),which may play an important role in the PPI network.Finally,the web tool Metascape was used to perform functional enrichment analysis of the core module.The results showed that 17 genes in the core module were mainly enriched in cross-chemical synaptic transmission,glutamatergic synaptic transmission,and positive regulation of synaptic transmission (Fig.5c).

Fig.5.Construction of protein-protein interaction (PPI) network and detection of key module.(a) PPI network of mutual DEGs in glioma.Circle size represents the significance of nodes in the network.(b) Core module of PPI network in glioma.(c) Results of functional enrichment analysis of core module in glioma performed using Metascape.

Fig.6.Identification and prognosis of hub genes in core module.(a) PPI network of ten hub DEGs in glioma.(b) Basic information of ten hub genes.The red-colored gene (GABRG2) indicates the highest degree of importance.(c) Prognosis of hub genes in glioma.P < 0.05 was considered as statistically significant.
3.4 Identification and survival analysis of key genes in the core module
The cytoHubba plug-in in Cytoscape was used to calculate the score of each gene in the core module,and the top 10 genes with the highest scores were selected as the key genes(Fig.6a).The results showed that 10 genes —GABRG2,SLC17A7,GRIN2A,SYNPR,RBFOX3,SYT1,RBFOX1,SLC12A5,GABRB2,and NEUROD6—were the key genes in the PPI network,among which GABRG2 had the highest degree of importance.The basic annotations of each gene and the MCC score are shown in Fig.6b.In order to further explore the impact of these genes on the prognosis of glioma patients,we analyzed the GEPIA database and discovered that these 10 low-expression genes were significantly related to the poor prognosis of glioma patients (Figs.6c and S3).
3.5 Gene expression analysis of GABRG2

Fig.7.Relative expression of GABRG2 and its potential molecular mechanism.(a) Relative expression of GABRG2 in 81 samples of normal brain tissue and 948 samples of glioma tissue (204 cases of WHO I,134 cases of WHO II,135 cases of WHO III,and 475 cases of WHO IV grade).(b) GABRG2 expression is downregulated in 20 glioma tissues compared with that in 15 normal brain tissues.(c) Relative expression of GABRG2 in glioma cell lines,primary cells,and HEB using qRT-PCR.(d) Potential molecular mechanisms of GABRG2 analyzed using GeneMANIA.****P < 0.0001,####P <0.0001,&&&&P < 0.0001.
Based on the basic information in these 17 GSE datasets,we divided the 948 glioma samples into four groups (204 cases of WHO I,134 cases of WHO II,135 cases of WHO III,and 475 cases of WHO IV).The results showed that the GABRG2 expression was significantly lower in gliomas than that in normal brain tissues.In addition,from WHO II to WHO IV,the GABRG2 expression was gradually downregulated with an increase in the pathological grade of the glioma (Fig.7a).However,the GABRG2 expression in WHO grade I gliomas was significantly lower than that in other grades of gliomas(Fig.7a).Most of the patients in this study with grade I gliomas were children whose brains had not completely developed.We further verified the GABRG2 expression in gliomas using the GEPIA and CGGA databases (Fig.S4a,b).qRT-PCR analysis demonstrated that GABRG2 expression was significantly downregulated in glioma tissues,primary cells,and glioma cell lines,which was consistent with our previous analyses (Fig.7b,c).In addition,analysis of the CGGA database also showed that the GABRG2 expression in IDH wild-type astrogliomas was significantly lower than that in mutants (Fig.S4c).Lastly,we used GeneMANIA software to analyze the biological process of GABRG2 and show the interactions between GABRG2 and 20 genes involved in GABA receptor complexes,ligand-gated anion channel activity,and GABA receptor activity (Fig.7d).Based on these results,we conclude that GABRG2 may affect the biological progression of glioma by affecting GABA receptor-related pathways.
4 Discussion
Glioma is the most common primary intracranial malignant tumor.Owing to its high heterogeneity,difficulty in complete resection,resistance to radiotherapy and chemotherapy,and ease of recurrence,the prognosis of patients with glioblastoma is abysmal.With proposal of the concept of precision medicine,molecular targeted therapy of glioma has attracted more attention.The challenge of identifying reliable molecular diagnostic markers has also become a focus of recent research in this area.With the continuous development of bioinformatics technology,microarray and deep sequencing have been widely used to explore gene-level changes,identify DEGs,and provide a comprehensive and in-depth platform for elucidating the relevant molecular mechanisms of tumor progression[38].
We selected and analyzed 17 GSE datasets that met our study criteria from the GEO database;81 samples of normal brain tissue and 948 samples of glioma tissue were included.R software and online tools were used to conduct bioinformatics analyses on each dataset.A total of 74 overlapping DEGs were identified.GO and KEGG analyses showed that all DEGs were mainly involved in GABAergic synaptic transmission,the γ-aminobutyric acid signaling pathway,and glutamatergic synapses,which reflected the complexity of glioma pathogenesis.Overall,the abnormal expression of multiple genes can affect the biological progression of glioma through different mechanisms.
To further screen out the key regulators of these mutual DEGs,we used the web tools STRING and Cytoscape to construct and analyze the PPI network and obtained a network composed of 17 nodes and 76 edges.GO analysis showed that the 17 genes were mainly enriched in the processes of crosschemical synaptic transmission,glutamatergic synaptic transmission,and brain development.Studies have shown that glutamatergic synaptic input to glioma cells drives brain tumor progression[39].Glioma cells integrate into communicating multicellular networks and are interconnected through neurite-like cellular protrusions[40].Further research is needed to clarify the exact molecular mechanisms of these pathways.In addition,we found that the top 10 hub genes in the core module were significantly correlated with the poor prognosis of glioma patients,indicating that the DEGs selected in this study may be potential biomarkers in the future.
Our research found that GABRG2,a key gene in the core module,has a significantly lower expression in glioma tissues in the 17 GSE datasets and TCGA and CGGA databases and was associated with the IDH classification of gliomas.After consulting the database,we found that GABRG2 was differentially expressed between different subtypes of diffuse astroglioma[41],which is also consistent with our reported results.However,the potential molecular mechanism by which GABRG2 affects the progression of glioma is yet to be discovered.GABRG2,which encodes the inhibitory neurotransmitter GABA receptor,can form functional GABA-gated chloride channels upon expressing alone in vitro[42],and its coding protein is the main component of the GABA synaptic channel[43].Previous studies have shown that GABRG2 is closely related to the pathogenesis of epilepsy,febrile seizures,and other diseases[44-46].Using GeneMANIA analysis,we found that GABRG2 participated in the process of GABA receptor activity.Studies have shown that GABAergic neurons regulate the proliferation of glioma cells in mice[47].Endogenous GABAA receptor activation leads to membrane depolarization,resulting in attenuated growth of low-grade glioma cells[48].Other studies have shown that tumor-associated seizures are a consequence of impaired GABAergic inhibition[49].GABA signaling and cellular chloride regulation processes are involved in oncogenesis and are associated with epileptic discharges in glioma patients[50].In summary,GABA and its related receptors play important roles in the occurrence of gliomas and the process of inducing epilepsy.GABRG2 may affect the progression of glioma by regulating GABA receptor-related pathways;nevertheless,further investigation of this mechanism is warranted.Our study provides a theoretical basis for future research on the correlation between GABRG2 and gliomas.However,indepth analyses of GABRG2 in the biogenesis of gliomas and more clinical data are needed to verify our results.In future research,we plan to clarify the molecular mechanism of GABRG2 in glioma and conduct clinical experiments targeting GABRG2 to alleviate glioma and its associated epilepsy.
Supporting information
The supporting information for this article can be found online at https://doi.org/10.52396/JUSTC-2022-0010.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81902525),USTC Research Funds of the Double First-Class Initiative (YD9100002002).
Conflict of interests
The authors declare that they have no conflict of interest.
Biographies
Xiaoming Zhangis currently pursuing a doctoral degree in Clinical Medicine at the University of Science and Technology of China.His research focuses on the role of non-coding RNA in pathogenesis of glioblastoma.
Shanshan Hureceived her Ph.D.degree in Cell Biology from the School of Life Sciences,University of Science and Technology of China.She is currently an associate research fellow at the First Affiliated Hospital of USTC,Division of Life Sciences and Medicine,University of Science and Technology of China.Her research interest involves GSC derived exosome in glioma-associated environment,non-coding RNA in GBM progression and evolution.

- 中国科学技术大学学报的其它文章
- IITZ-01 activates NLRP3 inflammasome by inducing mitochondrial damage
- Sphingosine-1-phosphate induces Ca2+ mobilization via TRPC6 channels in SH-SY5Y cells and hippocampal neurons
- Evolutionary game analysis of promoting the development of green logistics under government regulation
- Self-supervised human semantic parsing for video-based person re-identification
- The surrounding vehicles behavior prediction for intelligent vehicles based on Att-BiLSTM