
Emerging roles of GPR109A in regulation of neuroinflammation in neurological diseases and pain
2022-10-22 04:27KyleTaingLawrenceChenHanRongWeng
中国神经再生研究(英文版) 2023年4期
Kyle Taing, Lawrence Chen, Han-Rong Weng
Abstract Neuroinflammation plays a critical role in the pathological process of multiple neurological disorders and pathological pain conditions.GPR109A, a Gi protein-coupled receptor, has emerged as an important therapeutic target for controlling inflammation in various tissues and organs.In this review,we summarized current data about the role of GPR109A in neuroinflammation.Specifically, we focused on the pharmacological features of GPR109A and signaling pathways used by GPR109A to ameliorate neuroinflammation and symptoms in Alzheimer’s disease, Parkinson’s disease, multiple sclerosis, stroke, and pathological pain conditions.
Key Words: β-hydroxybutyrate; cytokines, HCAR2; lupus; neuroinflammation; neuropathic; niacin;nociception; synaptic
Introduction
Neuroinflammation, characterized by infiltration of leukocytes, activation of microglia and astrocytes, and overproduction of pro-inflammatory mediators in the nervous system, is a key pathological feature shared by numerous neurological diseases (DiSabato et al., 2016; Bachiller et al., 2018).Controlling neuroinflammation has become an important approach to the prevention and treatment of cell death in Alzheimer’s disease (AD) (Muzio et al., 2021; Ahmad et al., 2022), Parkinson’s disease (PD) (Badanjak et al., 2021; Muzio et al.,2021), multiple sclerosis (Thompson and Ciccarelli, 2020; Healy et al., 2022),and stroke (Ahmad et al., 2014; Tschoe et al., 2020).Neuroinflammation is also implicated in abnormal neuronal activation along with the pain signaling pathway in pathological pain conditions (Chen et al., 2018; Lacagnina et al.,2021).Targeting microglial receptors or signaling molecules has been proven to be a powerful means to control neuroinflammation in neurological diseases and chronic pain (Bachiller et al., 2018; Chen et al., 2018; Muzio et al., 2021).
Accounting for approximately 10-15% of all cells in the brain, microglia have long been known to act as macrophages, the first line of immune defense,within the central nervous system by actively surveying their surrounding microenvironment (Nimmerjahn et al., 2005; Bachiller et al., 2018; Chen et al.,2018; Muzio et al., 2021).Microglial cells are activated upon the stimulation of a host of pro-inflammatory receptors, including chemokine receptors(CX3CR1), purinergic receptors (P2X4R, P2X7R, P2Y12, P2Y13), Toll-like receptor 4 (Kobayashi et al., 2008; Taves et al., 2013; Tsuda et al., 2013; Grace et al., 2014), and colony-stimulating factor 1 receptor (Guan et al., 2016; Yan et al., 2017).Activation of such receptors results in the production of proinflammatory mediators and microglial proliferation (Kobayashi et al., 2008;Taves et al., 2013; Tsuda et al., 2013; Grace et al., 2014; Guan et al., 2016;Yan et al., 2017).Much less is known about the anti-inflammatory receptors expressed on microglial cells.Emerging studies indicate that G-protein-coupled receptor 109A (GPR109A), an anti-inflammatory G-inhibitory (G) proteincoupled receptor, regulates neuroinflammation in neurological diseases and chronic pain by regulating microglial activation.In this review, we will first introduce the history and pharmacological features of GPR109A.We will then discuss mechanisms and molecular pathways involving GPR109A within the microglia as well as how GPR109A may play a role in regulating neuroinflammation in neurological diseases and pathological pain conditions.
Search Strategy
PubMed and Google Scholar databases with default settings and no restrictions were used to search the literature in this review.“GPR109A” and each of its aliases [“Acid Receptor 2 (HCAR2)”, “Niacin Receptor 1 (NIACR1)”,“HM74a”, “HM74b”, and “PUMA-G”] were used as key words to search the literature regarding the nature of GPR109A on the databases.To obtain the literature specific to the roles of GPR109A in neurological disorders and pain, we searched the databases with “GPR109A” or one of its aliases [“Acid Receptor 2 (HCAR2)”, “Niacin Receptor 1 (NIACR1)”, “HM74a”, “HM74b”, and“PUMA-G”] in combination with “Alzheimer’s”, or “Parkinson’s”, or “multiple sclerosis”, or “stroke”, or “ischemia or ischemic”, or “Huntington’s”, or “epilepsy or epileptic”, or “neuropathy or neuropathic”, or “neuroinflammation or neuroinflammatory”, or “cytokine”, or “glial”, or “astrocyte”, or “microglia”,or “pain”.Since no literature was found regarding the role of GPR109A in Huntington’s disease or epilepsy, our review focuses on its role in Alzheimer’s disease, Parkinson’s disease, multiple sclerosis, stroke, and pathological pain conditions.
History and the Pharmacological Features of GPR109A
In 2003, three research groups independently discovered that GPR109A,an orphan G-protein-coupled receptor (GPCR), has a high affinity for niacin(also known as nicotinic acid and Vitamin B) (Soga et al., 2003; Tunaru et al., 2003b; Wise et al., 2003b; Offermanns et al., 2011).This receptor was deorphanized two years later when the endogenous ligand ketone body β-hydroxybutyrate (BHB) was found (Taggart et al., 2005).This receptor is also known as hydroxycarboxylic acid receptor 2 (HCAR2), niacin receptor 1 (NIACR1), HM74a, HM74b, and PUMA-G (Tunaru et al., 2003a; Chai et al., 2013).The gene coding GPR109A (HCAR2) is located on chromosome 12 (Band 12q24.31) in humans (Zellner et al., 2005).It has been shown in human embryonic kidney 293 (HEK293) cells that GPR109A forms either homo-dimers or hetero-dimers with GPR109B in both the plasma membrane and endoplasmic reticulum (Mandrika et al., 2010).GPR109B has more than 95% sequence homology with GPR109A with a difference in only 16 amino acids across their sequences (Wise et al., 2003b).The homo- or heterodimerization state is not altered by ligand binding.There is no significant difference in agonist-mediated cAMP signaling between cells with homo-and hetero-dimerization, and the dimerization is a constitutive process occurring early during biosynthesis (Mandrika et al., 2010).GPR109A embodies a highly conservative GPCR structure coupled with an inhibitory Gprotein (Wise et al.,2003a; Tuteja et al., 2017).GPR109A is localized to the cellular membranes of cells and expressed in multiple tissues and cell types (Table 1).
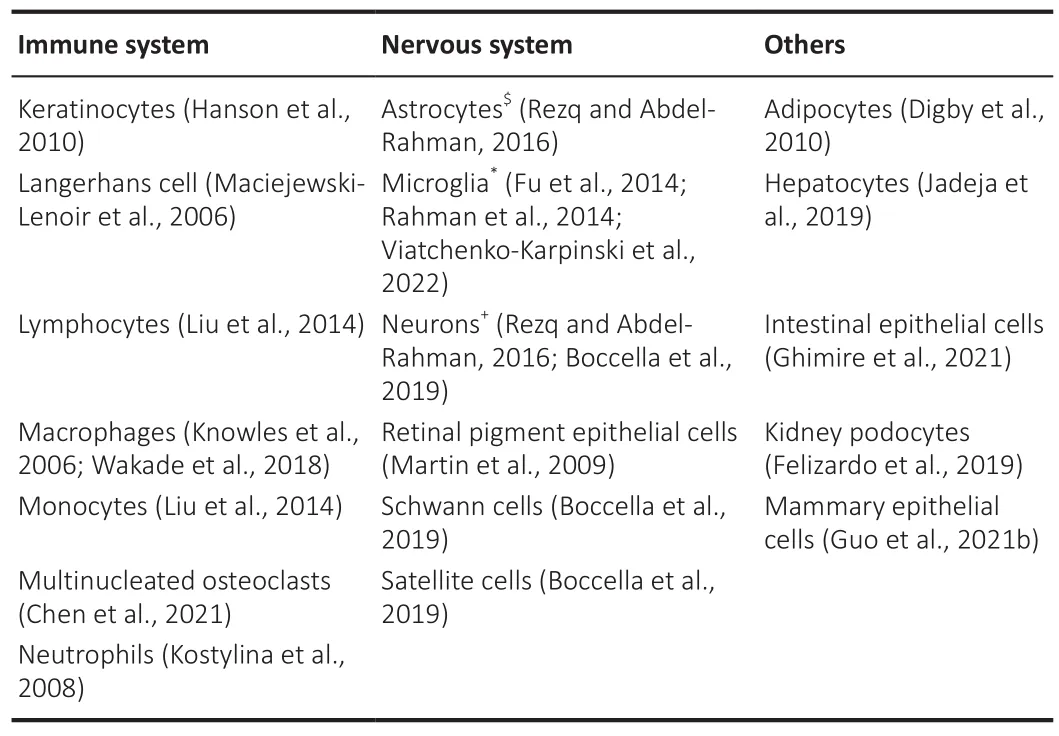
Table 1 |GPR109A expression in tissues and cellular types
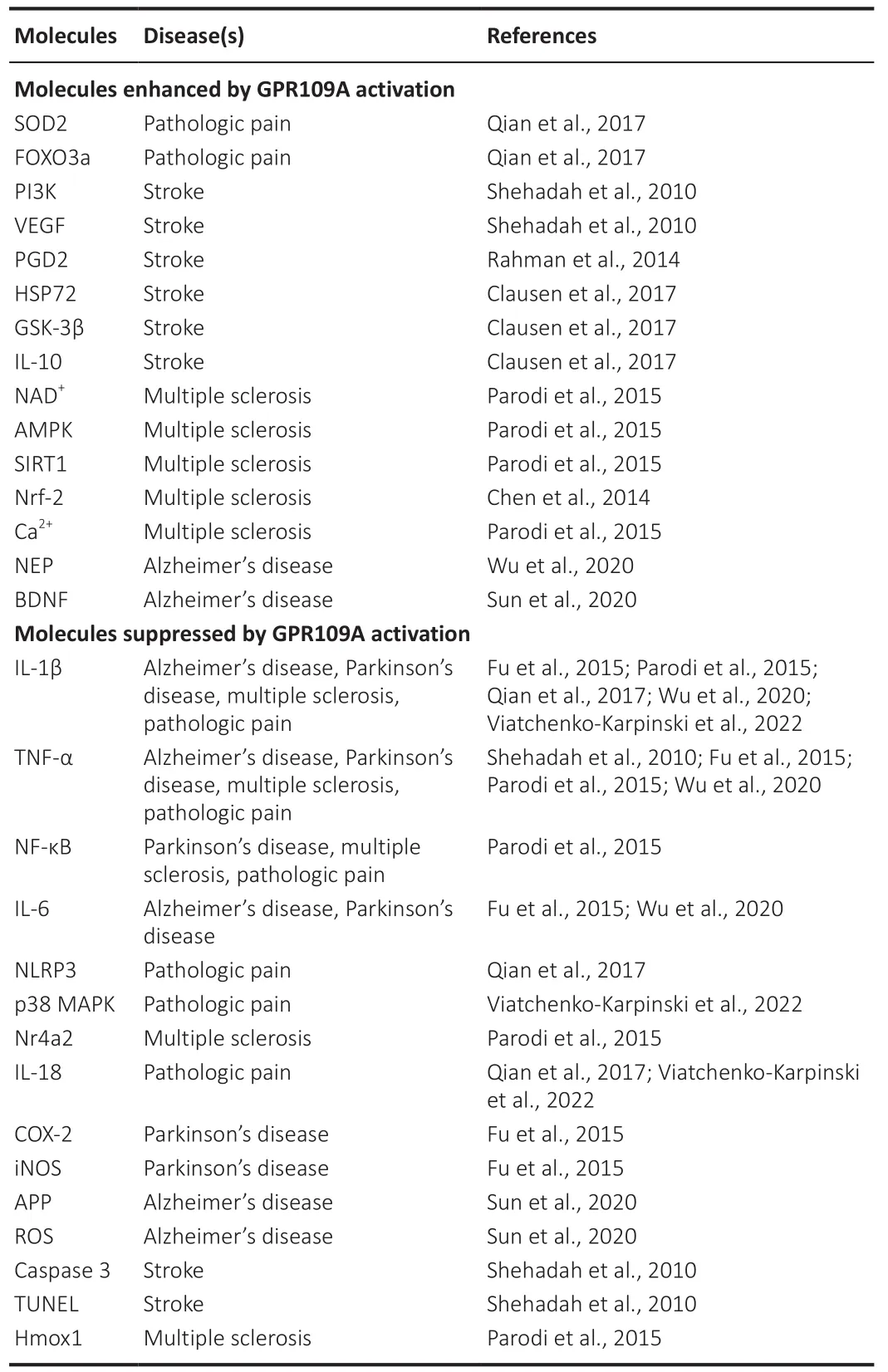
Table 2 |Summary of signaling molecules regulated by GPR109A
The most notable agonist for GPR109A is niacin, which lends its name to one of the receptor’s aliases (niacin receptor 1).The other endogenous agonists of GPR109A are BHB and butyrate, which are ketone bodies produced during ketosis (Tunaru et al., 2003a; Lee et al., 2020).BHB has a higher potency to GPR109A than butyrate (Taggart et al., 2005).The ECof BHB requiredfor the activation of GPR109A is approximately 700 μM (Gille et al., 2008)while the concentration of BHB is 44 μM in the cerebrospinal fluid and 62 μM in the blood under physiological conditions in rats (Wang et al., 2017).Thus, the concentration of BHB in the body under normal conditions is not sufficient to activate GPR109A.However, short-term starvation was found to induce activation of GPR109A due to elevated endogenous BHB levels through ketosis (Boccella et al., 2019), as ketone body levels rise in individuals undergoing fasting.BHB levels can be raised to approximately 5 mM concentration also by nutritional ketosis (Koppel and Swerdlow, 2018).It is noteworthy that niacin, BHB, and butyrate are not selective agonists for GPR109A.In addition to having a high-affinity for GPR109A, niacin also shows a low-affinity for GPR109B.Furthermore, niacin is an NADprecursor,which is required for over 500 enzymatic reactions and plays key roles in the regulation of mitochondrial metabolism, inflammation, meiosis, autophagy,and apoptosis (Rajman et al., 2018).Besides binding to GPR109A, butyrate and BHB respectively activate GPR41 and GPR43 (Brown et al., 2003; Won et al., 2013).BHB also inhibits class I and IIa histone deacetylases to regulate gene expression (Shimazu et al., 2013), and it acts as a metabolite to increase mitochondrial adenosine 5′-triphosphate production (Sato et al., 1995) and bolster antioxidant defenses (McCarty et al., 2015).
Synthetic agonists for GPR109A include drugs such as acipimox and acifran,both of which are niacin derivatives and hypolipidemic agents (Pike, 2005;Jung et al., 2007; Kamanna and Kashyap, 2007).Both acipimox and acifran have a high affinity for GPR109A and a low affinity for GPR109B (Kamanna and Kashyap, 2007).Monomethyl fumarate (MMF), an active metabolite of dimethyl fumarate (DMF), has been demonstrated to be an agonist of GPR109A (Hanson et al., 2010; Venci and Gandhi, 2013).More recent synthetic selective GPR109A agonists include MK-1903, which was developed for the treatment of dyslipidemia (Boatman et al., 2012), and GSK2560743,which reduces glucose levels in patients with type 2 diabetes (Dobbins et al.,2013).Finally, mepenzolate bromide, a synthetic antimuscarinic drug, is a GPR109A receptor blocker (Singh et al., 2012).
Roles of GPR109A in Neurological Diseases
GPR109A was originally recognized to perform three key functions in the cells expressing this receptor: (1) inhibiting lipolysis (breakdown of fats) in adipocytes (Plaisance et al., 2009), (2) mediating niacin-induced apoptosis in mature neutrophils (Kostylina et al., 2008), and (3) suppressing atherogenesis in arteries (Digby et al., 2012; Chai et al., 2013; Zhang et al., 2021).Moreover,this receptor is also implicated in mitigating many other pathophysiological processes, including the reduction of chemokine and proinflammatory cytokine production (Digby et al., 2012), suppression of mammary tumorigenesis by inhibiting cancer cell survival (Elangovan et al., 2014), and attenuation of colonic inflammation and carcinogenesis by communicating with the gut microbiome (Thangaraju et al., 2009).The beneficial effects induced by activation of microglial GPR109A in neuroinflammation-related neurological diseases, such as Alzheimer’s disease, PD, stroke, multiple sclerosis, and pathological pain conditions, have been revealed in recent years.
Alzheimer’s disease
AD patients present with the progressive and irreversible decline of cognitive,behavioral, and sensorimotor functions (Knopman et al., 2021).AD is pathologically characterized by the accumulation of extracellular deposits of insoluble amyloid-β (Aβ) and intracellular aggregates of hyperphosphorylated tau (Knopman et al., 2021).Although there is currently no cure for AD, studies have shown that this disease is correlated with increased levels of microglial and astrocytic activation and increased production of proinflammatory cytokines (Knopman et al., 2021; Leng and Edison, 2021; De Sousa, 2022).Activating GPR109A with ketone bodies was reported to produce protective effects in animal models andin vitro
culture cells.In the 5XFAD AD mouse model, BHB treatment significantly improves passive avoidance behaviors and responses to the Morris water maze test and nest construction (Wu et al.,2020).These are accompanied by attenuation of Aβ accumulation and senile plaques in the brain.Microglial activation and the production of interleukin 1 beta (IL-1β), tumor necrosis factor-alpha (TNF-α), and interleukin 6 (IL-6) in the brain are also reduced (Wu et al., 2020).BHB suppresses the expression of amyloid precursor protein (APP) but increases the amount of neprilysin(NEP), a degradation enzyme for Aβ (Wu et al., 2020).In GPR109A knockout mice, BHB failed to modulate APP and NEP levels (Wu et al., 2020).Furthermore, the authors found that HT22 hippocampal cells treated with BHB have an improved mitochondrial respiratory function and increased adenosine 5′-triphosphate production.The beneficial effects of GPR109A were further demonstrated inin vitro
mouse neuroblastoma N2a cells (Sun et al., 2020).In this study, sodium butyrate was used to activate GPR109A.The authors found that sodium butyrate produces protective effects on Aβinduced N2a cell injury.APP levels and production of Aβ-induced reactive oxygen species (ROS) are suppressed in cells treated with sodium butyrate.Sodium butyrate treatment increases the amount of NEP, GPR109A, and brain-derived neurotrophic factor (BDNF), a member of the neurotrophin family that confers protective effects against AD.These effects are attenuated when the Gi protein is inhibited by pertussis toxin, consistent with the nature of Gi GPCRs (Sun et al., 2020).Together, these findings strongly suggest GPR109A agonists as a promising scaffold for therapeutic development.Parkinson’s disease
PD clinically manifests with tremors, rigidity, gait abnormalities, and late-onset cognitive dysfunction.The PD pathology is characterized by dopamine deficiency due to loss of neurons in the substantia nigra as well as intracellular alpha-synuclein aggregation (Poewe et al., 2017).Microglial activation in the substantia nigra was found in post-mortem studies(Hartmann, 2004).It is believed that the pathogenesis of PD may involve various mechanisms, including oxidative stress, mitochondrial dysfunction,and neuroinflammation (Muzio et al., 2021).Recent studies have reported the beneficial effects of BHB and niacin in the management of PD in animal models and clinical studies.Specifically, BHB improves the motor dysfunction and dopamine depletion of the PD rat model induced by intranigral injection of lipopolysaccharide (LPS) (Fu et al., 2015).These beneficial effects are accompanied by the inhibition of microglial over-activation as well as the protection of dopaminergic neurons in the substantia nigra (Fu et al., 2015).In primarily mesencephalic mixed neuron-glial cultures, activation of GPR109A with BHB attenuates microglial activation and levels of phosphorylated nuclear factor kappa B (NF-κB) induced by LPS, which, in turn, dampens the LPS-induced protein and mRNA expression of various pro-inflammatory mediators [e.g., inducible nitric oxide synthase, cyclooxygenase-2 (COX-2),TNF-α, IL-1β, and IL-6] (Fu et al., 2015).Activation of GPR019A with niacin reduces the production of the pro-inflammatory cytokines IL-1β and IL-6 induced by LPS in BALB/c murine macrophage RAW264.7 cells (Giri et al.,2019).Niacin produces this anti-inflammatory effect by suppressing the nuclear translocation of phosphorylated NF-κB induced by LPS (Giri et al.,2019).Interestingly, PD patients receiving niacin supplements have a higher ratio of M2:M1 macrophages as well as improved quality of life (Wakade et al., 2018).Further study on the changes in molecular signaling pathways in patients receiving niacin would provide a rationale for the application of niacin in PD treatment.
Multiple sclerosis
Multiple sclerosis (MS) is autoimmunity-initiated inflammatory demyelination of the central nervous system, commonly presenting with various neurologic symptoms (Healy et al., 2022).Most treatments are disease-modifying agents aimed to decrease the likelihood of relapses and delay the progression of neurodegeneration.A recent study has recognized that activation of glial cells plays an important role in the disease progressive process (Healy et al., 2022).One specific medicine approved by the FDA in 2013 for treating patients with relapsing MS is dimethyl fumarate (DMF) (Venci and Gandhi,2013), which was previously used to treat psoriasis (Venci and Gandhi, 2013).DMF is converted to its active metabolite, monomethyl fumarate (MMF),which is known to be a nicotinic receptor agonist (Tang et al., 2008; Hanson et al., 2010).Although the mechanisms underlying the beneficial effects induced by DMF remain not clearly understood, it was demonstrated that GPR109A at least in part mediates the beneficial effects of MMF.In mice with experimental autoimmune encephalomyelitis, a murine model of MS,Chen’s group in 2014 reported that DMF treatment improves motor function,reduces immune cell infiltration, and decreases demyelination of the spinal cord in wild-type mice.These beneficial effects are abolished in GPR109A knockout mice, providing evidence for the dependence of DMF on GPR109A(Chen et al., 2014).In murine microglial cell line N9, Parodi et al showed that through activation of GPR109A, MMF switches the LPS-activated microglia from a classically activated, pro-inflammatory type to an alternatively activated, neuroprotective one (Parodi et al., 2015).MMF elevates Calevels and increases the activity of AMPK (phosphorylated AMPK) in microglia.The authors demonstrated that AMPK activation results in the activation of sirtuin 1 (SIRT1) via NADgeneration.SIRT1, in turn, inhibits NF-κB via acetylation and suppresses the production of pro-inflammatory molecules such as TNF-α, IL-1β, and heme oxygenase 1, and nuclear receptor subfamily 4 group A member 2 (Parodi et al., 2015).At the synaptic levels, DMF treatment normalizes the increased frequency of excitatory postsynaptic currents in corticostriatalneurons in mice with EAE (Parodi et al., 2015).MMF reduces monocyte transendothelial migration and adhesion to inflamed human brain endothelial cells (Lim et al., 2016).Activation of mRNA nuclear factor (erythroid-derived)related factor-2 (Nrf2) has been associated with mechanisms of action of DMF/MMF with discrepant reports.DMF was found to produce protective effects on oligodendrocytes, myelin, axons, and neurons in mice with EAE,and reduce oxidative stress via activation of the Nrf2 antioxidant pathway(Chen et al., 2014).However, another study later found in the EAE model that DMF treatment ameliorates disease signs in Nrf2 knockout mice, both clinically and histologically (Schulze-Topphoff et al., 2016).DMF treatment did not change the total count of CD4T cells in either WT or Nrf2 knockout mice,but it did lower the amount of both Th1 and Th17 cells in both mice models(Schulze-Topphoff et al., 2016).Further investigation is required to resolve such discrepancies.
Stroke
Stroke refers to a blockage or bleeding of the blood vessels that either interrupts or reduces the supply of blood to the brain.After acute stroke episodes, neuroinflammation further injures neurons, glia, vascular cells, and extracellular matrix scaffolding (Jayaraj et al., 2019).In an ischemic mouse model induced by the distal middle cerebral artery occlusion, it has been shown that infarcts in mice on ketogenic diets or receiving BHB or nicotinic acid are significantly smaller than those in control groups (Rahman et al.,2014).More importantly, this study demonstrated that activation of GPR109A on monocytes and/or macrophages produces such protective effects via the production of prostaglandin D2 (PGD2) by cyclooxygenase 1 (COX-1) and the hematopoietic PGD2 synthase (Rahman et al., 2014).The beneficial role of GPR109A in stroke is in line with other studies where the effects of BHB,niacin or DMF/MMF were tested on animal middle cerebral artery occlusion stroke models.It was reported that intraventricular injection of BHB improves neurological scores and reduces infarct volume after ischemia via upregulating the BHB transporter (sodium-coupled monocarboxylate transporter 1) and activating the extracellular-signal-regulated kinase/cyclic AMP-responsive element-binding protein/endothelial nitric oxide synthase pathway (Li et al.,2021a).Intravenous injection of BHB significantly reduces cerebral infarct area, edema formation, lipid peroxidation, and neurological deficits (Suzuki et al., 2002).Animals treated with Niaspan, an extended-release formulation of niacin, have smaller infarct areas and improved functional recovery after stroke (Shehadah et al., 2010).Niaspan treatment reduces levels of apoptotic markers (TUNEL and cleaved caspase-3) and TNFα but raises levels of vascular endothelial growth factor and phosphorylated phosphatidylinositol 3-kinase in the ischemic brain (Shehadah et al., 2010).Synaptic plasticity and neuronal axon growth are also promoted by niacin treatment (Cui et al., 2010).Many studies have shown that DMF/MMF ameliorates areas of infarct and functional recovery in mice with middle cerebral artery occlusion (Safari et al., 2019; Li et al., 2021b).DMF and its breakdown product, MMF, alleviate oxidative stress and inflammation after ischemic stroke (Lin et al., 2016;Safari et al., 2019; Li et al., 2021b).Such protective effects are associated with suppression of glial activation (Yao et al., 2016) and glycogen synthase kinase-3β expression, and increased expression of heat shock protein 72 and interleukin 10 (IL-10), an anti-inflammatory cytokine (Clausen et al., 2017).Nrf2 is suggested to mediate the anti-oxidative and anti-inflammatory role induced by DMF/MMF (Zhao et al., 2015; Lin et al., 2016; Yao et al., 2016;Safari et al., 2019; Li et al., 2021b).Interestingly, GPR109A mRNA levels were elevated in both the treated and untreated groups, suggesting that the upregulation in GPR109A does not come with MMF therapy but with the initial stroke itself (Clausen et al., 2017).
Roles of GPR109A in Pathological Pain
Pain is one of the most common reasons people seek medical treatment,as well as a leading cause of disability and disease burden globally (Disease et al., 2018).Clinical management of chronic pain remains a challenge because currently available analgesics (including NSAIDs, antidepressants,anticonvulsants, and opioids) (Israel et al., 2021) are neither potent nor safe(Gregus et al., 2021; Israel et al., 2021).Accordingly, the development of novel analgesics with high potency and safety features is in great demand.It is widely accepted that pathologic pain occurs when neurons along the pain signaling pathway are abnormally activated.Neurons in the spinal dorsal horn are part of the pain signaling pathway.Excessive activation of spinal dorsal horn neurons,termed spinal central sensitization, is known to play a critical role in the genesis of pathologic pain (Woolf, 2011).Studies over the past two decades have demonstrated that neuroinflammation greatly modulates neuronal activities in the spinal dorsal horn.Glial cells can enhance neuronal activities through releasing pro-inflammatory cytokines [such as TNF-α (Kawasaki et al., 2008),IL-1β (Kawasaki et al., 2008; Yan and Weng, 2013), IL-6 (Kawasaki et al., 2008),IL-18 (Viatchenko-Karpinski et al., 2022)], and chemokines (Cao et al., 2014;Chen et al., 2018), as well as reducing glial glutamate transporter activity (Yan et al., 2014).For example, IL-1β enhances spinal excitatory synaptic activity by increasing presynaptic glutamate release (Yan and Weng, 2013), postsynaptic AMPA (Yan and Weng, 2013), and NMDA receptor activity (Kawasaki et al.,2008), as well as reducing glial glutamate transporter activity (Yan et al., 2014).In addition, IL-1β also suppresses GABAergic inhibitory synaptic activity by reducing both presynaptic synthesis of GABA transmitter and postsynaptic GABA receptor activity (Yan et al., 2015).Glial activation and production of pro-inflammatory mediators are controlled by either receptors on the cell plasma membrane or intracellular signaling molecules in microglia.GPR109A has emerged as an important target for regulating neuroinflammation in the pain signaling pathway and conquering chronic pain (Boccella et al., 2019;Viatchenko-Karpinski et al., 2022).
Studies in recent years have shown that the administration of DMF and BHB,both GPR109A agonists, in animals produces analgesic effects.Oral DMF administration strongly prevents thermal and mechanical hypersensitivity induced by tibial bone fracture (Guo et al., 2021a), intervertebral disc degeneration (Zhu et al., 2020), complete Freund’s adjuvant-induced arthritis(Lal et al., 2021), and nerve injury (Li et al., 2020).Mechanistically, DMF treatment blocks the increased protein expression of IL-1β and IL-6 in the hind paw skin of mice with a bone fracture (Guo et al., 2021a).In rats with complete Freund’s adjuvant-induced arthritis, oral treatment of DMF reduces protein levels of IL-1β, TNF-α, COX-2, and NF-κb in the joint tissue (Lal et al.,2021).Protein expression levels of IL-1β and monocyte chemoattractant protein-1 in the dorsal root ganglion in rats with nerve injury are attenuated by DMF treatment (Li et al., 2020).In Nrf2 knockout mice, the beneficial effects induced by DMF in chronic pain induced by the tibial bone fracture are abolished (Guo et al., 2021a).Moreover, Nrf2 is considered to mediate the antioxidant and anti-inflammatory signaling by DMF in the complete Freund’s adjuvant-induced arthritis (Lal et al., 2021) and neuropathic pain (Li et al.,2020).
Qian et al.(2017) reported that BHB treatment improves functional recovery and neuropathic pain in mice with spinal cord injuries.BHB was continuously administered to mice via a subcutaneous mini-osmotic pump following a mid-thoracic spinal contusion injury at the T9-T10 level.BHB Treatment significantly improves locomotor function and attenuates mechanical and thermal hypersensitivity induced by spinal cord injury (Qian et al., 2017).Mechanistically, BHB treatment prevents loss of motor neuron function and suppresses microglia activation induced by spinal cord injury via (1)enhancing histone acetylation and protein expression of the transcription factor Forkhead box class O 3a, catalase, and superoxide dismutase 2; (2)suppressing the nucleotide-binding oligomerization domain-like receptor pyrin domain containing 3 inflammasome activation; (3) reducing IL-1β and IL-18 cytokine levels; and (4) ameliorating mitochondrial function and oxidative stress (Qian et al., 2017).The authors reported that BHB concentration in the spinal cord in mice receiving BHB via a mini-osmotic pump reaches a level of approximately 1.6 mmol (Qian et al., 2017), a concentration that is higher than the EC(approximately 700 μmol) for BHB to activate GPR109A.Although the authors in their report did not study the involvement of GPR109A in experiments, it is conceivable that activation of GPR109A at least mediates in part the effects induced by BHB in their experiments.
Recently, the role of GPR109A in the regulation of pathological pain was directly addressed in mice with neuropathic pain and mice with chronic pain caused by systemic lupus erythematosus (SLE).At the periphery, GPR109A was found in Schwann cells in the sciatic nerve, and neurons and satellite cells in the dorsal root ganglia (Boccella et al., 2019).In the spinal dorsal horn, GPR109A is expressed in spinal microglia but not in astrocytes or neurons (Viatchenko-Karpinski et al., 2022).GPR109A protein expression is significantly increased in the dorsal root ganglia in mice with peripheral nerve injury, and in the spinal dorsal horn in mice with chronic pain caused by SLE(Viatchenko-Karpinski et al., 2022).In mice with neuropathic pain induced by chronic constriction of the sciatic nerve or spared nerve injury, it was reported that single and repeated oral administration of DMF attenuates preexisting mechanical allodynia in both male and female mice (Boccella et al.,2019).BHB given intraperitoneally or raised by 60-hour starvation in mice also produces similar effects.These effects are abolished in GPR109A knockout mice, indicating that GPR109A mediates the analgesic effects induced by DMF and BHB on chronic pain induced by nerve injury (Boccella et al., 2019).More recently, we defined the role of GPR109A in chronic pain induced by SLE in MRL/lpr (lupus-prone) mice, a SLE mouse model.MRL/lpr mice spontaneously develop chronic pain at the age of 11 weeks and older (Yan et al., 2017; Viatchenko-Karpinski et al., 2022).This is concurrently accompanied by increased activation of microglia and astrocytes, as well as increased p38 mitogen-activated protein (MAP) kinase activity and production of IL-1β and IL-18 in the spinal dorsal horn (Yan et al., 2017; Viatchenko-Karpinski et al., 2022).At the synaptic level, mice with chronic pain induced by SLE have increased glutamate release from primary afferent terminals (Viatchenko-Karpinski et al., 2022) and reduced activity and protein expression of glial glutamate transporters (Yan et al., 2017) in the spinal dorsal horn.It has been demonstrated that IL-18 enhances glutamate release from the presynaptic terminals (Viatchenko-Karpinski et al., 2022) while IL-1β leads to suppression of glial glutamate transporter activity in the spinal dorsal horn (Yan et al.,2017).Intrathecal administration of a GPR109A specific agonist (MK1903)attenuates chronic pain induced by SLE in the male and female SLE mouse model (Viatchenko-Karpinski et al., 2022).The enhanced activity of p38 MAP kinase in mice with chronic pain is suppressed upon activation of GPR109A receptors in the microglia.Furthermore, activation of GPR109A receptors reduces the activity of cathepsin B and the production of mature IL-1β and interleukin-18 (IL-18) in the spinal dorsal horn in MRL/lpr mice with chronic pain (Viatchenko-Karpinski et al., 2022).Cathepsin B is critically engaged in the production of mature IL-1β and IL-18 from pro-IL-1β and IL-18 (Sun et al.,2012).These findings indicate that activation of spinal GPR109A produces analgesic effects in MRL/lpr mice with chronic pain through suppressing p38 MAP Kinase activity, mitigating production of IL-18 and IL-1β, and reducing glutamatergic activity at the spinal dorsal horn (Figure 1).Further investigation into the molecular pathways used by GPR109A to suppress the production of pro-inflammatory cytokines and the molecular pathways regulating the protein expression of GPR109A is warranted.
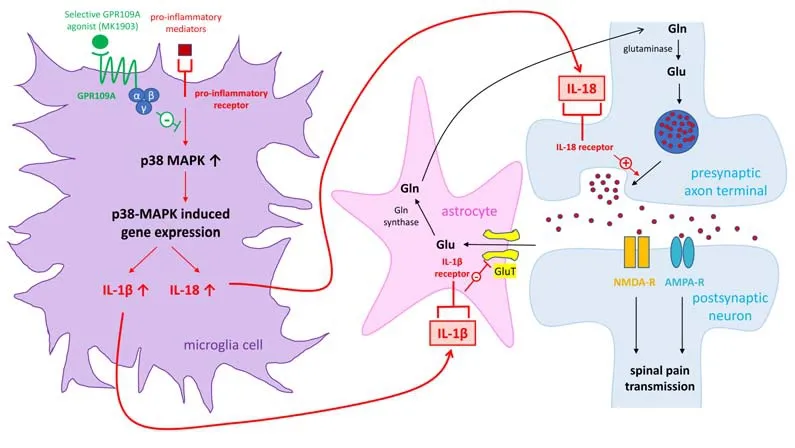
Figure 1|Molecular signaling pathways used by GPR109A to attenuate spinal neuroinflammation and chronic pain induced by SLE in mice.
Concluding Remarks
Neuroinflammation is a critical pathological process implicated in multiple neurological disorders including Alzheimer’s disease, Parkinson’s disease,multiple sclerosis, stroke, and pathological pain conditions.Accumulating data have suggested that activation of GPR109A receptors ameliorates symptoms in these neurological disorders and chronic pain via suppressing pro-inflammatory signaling pathways and the production of pro-inflammatory mediators as well as enhancing anti-inflammatory signaling pathways (Table 2).The majority of available data on the role of GPR109A in neuroinflammation were derived from experiments where non-specific GPR109 agonists (such as niacin, BHB,and MMF) were used in combination with GPR109A knockout techniques to determine the contribution of GPR109A signaling.Upon administration of non-specific agonists, however, the compounding effects induced by signaling pathways other than the GPR109A signaling cannot be ruled out.Future studies to understand the in-depth biology of GPR109A will open new avenues to identify and develop novel therapeutic targets for the treatment of neuroinflammation-related neurological diseases and chronic pain.
Author contributions:
Conception and design: HRW; literature search and manuscript writing: KT, LC, and HRW.All authors approved the final manuscript.
Conflicts of interest:
The authors declare no conflicts of interest.
Open access statement:
This is an open access journal, and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.

- 中国神经再生研究(英文版)的其它文章
- Potential physiological and pathological roles for axonal ryanodine receptors
- Multi-targeted anti-inflammatory drugs for the treatment of neurological disorders
- Melatonin, tunneling nanotubes, mesenchymal cells,and tissue regeneration
- MicroRNAs as potential biomarkers in temporal lobe epilepsy and mesial temporal lobe epilepsy
- Notice of Retraction
- Roles of constitutively secreted extracellular chaperones in neuronal cell repair and regeneration