
Potential physiological and pathological roles for axonal ryanodine receptors
2022-10-22 04:27DavidStirling
中国神经再生研究(英文版) 2023年4期
David P.Stirling
Abstract Clinical disability following trauma or disease to the spinal cord often involves the loss of vital white matter elements including axons and glia.Although excessive Ca2+ is an established driver of axonal degeneration, therapeutically targeting externally sourced Ca2+ to date has had limited success in both basic and clinical studies.Contributing factors that may underlie this limited success include the complexity of the many potential sources of Ca2+ entry and the discovery that axons also contain substantial amounts of stored Ca2+ that if inappropriately released could contribute to axonal demise.Axonal Ca2+ storage is largely accomplished by the axoplasmic reticulum that is part of a continuous network of the endoplasmic reticulum that provides a major sink and source of intracellular Ca2+from the tips of dendrites to axonal terminals.This “neuron-within-a-neuron” is positioned to rapidly respond to diverse external and internal stimuli by amplifying cytosolic Ca2+ levels and generating short and long distance regenerative Ca2+ waves through Ca2+ induced Ca2+ release.This review provides a glimpse into the molecular machinery that has been implicated in regulating ryanodine receptor mediated Ca2+ release in axons and how dysregulation and/or overstimulation of these internodal axonal signaling nanocomplexes may directly contribute to Ca2+-dependent axonal demise.Neuronal ryanodine receptors expressed in dendrites, soma, and axonal terminals have been implicated in synaptic transmission and synaptic plasticity, but a physiological role for internodal localized ryanodine receptors remains largely obscure.Plausible physiological roles for internodal ryanodine receptors and such an elaborate internodal binary membrane signaling network in axons will also be discussed.
Key Words: axomyelinic synapse; axon; axoplasmic reticulum; calcium; ryanodine receptor; secondary axonal degeneration; spinal cord injury; voltage-gated calcium channel; white matter injury
Background
Since the remarkable discovery that ryanodine receptor (RyR) mediated Carelease from internal Castores can contribute to central myelinated fiber conduction failure following white matter injury (Thorell et al., 2002;Ouardouz et al., 2003), much has been learned about the molecular mechanisms that regulate the release and storage of internal Cawithin axons and their supporting glia.Resting levels of free Cawithin the neuronal soma, dendrites, and axon must be tightly regulated to avoid Camediated damage but be readily available for the intraneuronal signaling needs of the cell that often expands over great distances.This task is largely accomplished through a continuous network of endoplasmic reticulum (ER) that extends from the tips of dendrites to axonal terminals providing a major sink and source of intracellular Ca.This excitable and dynamic intracellular network presumably functions as a “neuron-within-a-neuron” and is positioned to rapidly respond to diverse external and internal stimuli by amplifying cytosolic Calevels and generating short and long distance regenerative Cawaves (Berridge, 1998).This review provides a glimpse into the molecular machinery that has been implicated in RyR-mediated Carelease in axons and how dysregulation and/or overstimulation of these internodal axonal signaling nanocomplexes may directly contribute to Ca-dependent axonal demise.Plausible physiological roles for such an elaborate internodal binary membrane signaling network will also be discussed.
Search Strategy and Selection Criteria
PubMed search was performed using keywords ryanodine receptor AND axon up to May 24, 2022.
Physiological Roles for Neuronal Ryanodine Receptor-Mediated Ca2+ Release
The rapid and regenerative release of Cafrom the ER lumen is mediated in part by RyR.There are three known isoforms (RyR1-3) and RyR1 and RyR2 are best known for their established role in excitation-contraction coupling in skeletal and cardiac muscle respectively.In skeletal muscle, RyR1 is activated by physical interactions when the voltage-gated L-type Cachannel Ca1.1 undergoes conformational changes following depolarization of the sarcolemma (Woll and Van Petegem, 2022).Thus, RyR1 activation occurs without the need for an initial Cainflux.In contrast, in cardiac muscle, RyR2 is activated by Cainflux following activation of the voltage-gated L-type Cachannel Ca1.2 (Woll and Van Petegem, 2022).RyR can then stimulate other RyR propagating the Casignal through a process termed Cainduced Carelease (CICR).
All three isoforms are expressed in the brain with RyR2 being the most abundant (Abu-Omar et al., 2018; Santulli et al., 2018).RyR1 has been detected in most brain regions but is highly expressed in the cerebellum within Purkinje cells, and the dentate gyrus of the hippocampus (Furuichi et al., 1994).RyR2 is also widely distributed with the highest levels of expression within the hippocampus CA1-3 regions and dentate gyrus, the granule cell layer of the cerebellum, and cerebral cortex (Sah et al., 1993; Furuichi et al., 1994).RyR3 expression often overlaps the other RyR isoforms with high expression levels within the CA1 region of the hippocampus (Sah et al., 1993;Furuichi et al., 1994).RyR is exclusively localized to the ER and axoplasmic reticulum (AR) and have been detected in dendritic shafts, spines, soma,axonal initial segment, axon, and axonal terminals (Segal and Korkotian, 2014;Abu-Omar et al., 2018).
Consistent with their differential subcellular distribution and expression within specific regions of the brain, RyR has been implicated in synaptic transmission, synaptic plasticity, learning, and memory (Abu-Omar et al.,2018; Santulli et al., 2018; Woll and Van Petegem, 2022).In support, RyR-mediated Carelease plays an important role in long-term potentiation and long-term depression, two activity-dependent forms of synaptic plasticity that are commonly used to investigate the cellular basis of learning and memory formation (Baker et al., 2013; Paula-Lima et al., 2014).For example, recordings of presynaptic Catransients at the hippocampal mossy fiber-CA3 synapse revealed that CICR mediated by RyR amplified presynaptic Calevels in an activity-dependent manner which is critically important for presynaptic forms of plasticity (Shimizu et al., 2008).Interestingly, RyR2 expression was localizedspecifically within the mossy fiber axons rather than their terminals, whereas RyR1 was expressed postsynaptically.Thus, the differential subcellular location of the RyR isoforms provides important insight into their role in neuronal Casignaling, function, and dysfunction.
More recently, a novel mouse model of GFP-tagged RyR2 was used to more definitively determine the subcellular localization of RyR2 within the hippocampus and revealed that RyR2 was localized to the soma and dendrites of CA1 pyramidal neurons, dentate gyrus granular neurons, and mossy fiber axons, but absent from dendritic spines (Hiess et al., 2022).Utilizing a gain of function RyR2 mutant mouse model, it was demonstrated that RyR2-mediated Carelease plays an important role in neuronal excitability, learning, and memory (Hiess et al., 2022).RyR has also been shown to propagate the Casignal generated in dendritic spines, dendrites, and soma to the nucleus to induce known gene expression changes required for learning and memory(Lobos et al., 2021).
In contrast to RyR2 and RyR1, RyR3 knockout mice are viable and therefore their behavior, as well as their function, can be assessed.Compared to littermate controls, RyR3 knockout mice were generally hyperactive, less social, exhibited impairments in contextual fear conditioning and passive avoidance, and had altered spatial learning.However, discrepancies with spatial and working memory as well as reversal learning performance were reported between the studies (Takeshima et al., 1996; Balschun et al., 1999; Matsuo et al., 2009).In agreement with the hippocampal associated behavioral differences, RyR3 knockout mice had altered long-term potentiation and LDP (Balschun et al., 1999; Futatsugi et al., 1999; Shimuta et al., 2001).More recently, it was shown that RyR3 activity and CICR play an essential role in neuronal excitability through modulation of activitydependent potentiation of slow after hyperpolarising current involved in spike frequency adaptation (Tedoldi et al., 2020) adding to the complexity of RyR3 in neuronal function.
The generation of conditional neuron-specific RyR1, -2, and -3 mice will help clarify the precise physiological roles of each of the RyR isoforms in the nervous system.Collectively the data support an important physiological role for RyR in synaptic plasticity, learning, and memory.Given the importance of RyR in these aspects of neuronal function, it is not surprising that dysregulation of neuronal RyR mediated Carelease through trauma or disease could contribute to neuronal hyperexcitability, neurodegeneration,cognitive impairment, and white matter injury.As a full exploration of the role of RyR in developmental disorders, aging, and neurodegenerative disease are beyond the scope of this review the interested reader is referred to additional articles on these important areas of research (Abu-Omar et al., 2018; Kushnir et al., 2018; Keil et al., 2019; Sethi et al., 2021).
Ca2+ and White Matter Injury
Central myelinated axons provide critical communication between neurons,and disruption through injury or disease contributes to serious and often permanent clinical disability.Although the precise molecular mechanisms that mediate axonal injury are incompletely understood, it is widely accepted that axonal degeneration is set in motion by an influx of extracellular Cathat if unabated culminates in Ca-dependent enzymatic cleavage events,cytoskeletal disruption, and eventual axonal demise (Hill et al., 2016).
Mechanistically,ex vivo
preparations of white matter (e.g., optic nerve, and dorsal columns) have been instrumental in our understanding of how axons are injured by a variety of insults (Stirling and Stys, 2010).Advantages of these models include the ability to maintain highly controlled environmental conditions to mimicin vivo
conditions but be able to precisely manipulate the external milieu (e.g., removal of external Cafrom the perfusate).These models when combined with electrophysiological and optical imaging recordings of cell type specific fluorescent markers and genetically encoded Caindicators/dyes, provide a unique window into white matter function,and the dynamic response of axons and their supportive glia to injury in real time.Althoughex vivo
preparations closely mimic what is observed inin vivo
models, limitations clearly exist including a relatively short period of observation and the exclusion of blood-derived elements.“Outside in” and “Inside out” Sources of Ca2+
Important insights gained from usingex vivo
white matter preparations have shown that removal of external Cafrom the perfusate is highly protective in several models of white matter injury, establishing Caentry from external sources as detrimental (Stys et al., 1990; Stirling and Stys, 2010).Subsequent pharmacological studies revealed that voltage-gated Cachannels, reverse operation of the Na-Caexchanger, and glutamate receptors accounted for the majority of Cainflux (Fern et al., 2014).In addition to Cainflux across the axolemma, Cacan also be mobilized from intra-axonal Castores.The major Castore in axons is the continuous network of tubular smooth ER referred to as the AR and to a lesser extent mitochondria.Indeed, total axonal Calevels approach 1 mM in axons, and free axoplasmic levels of Caare~100 nM, suggestive that inappropriate and excessive release of Cafrom the AR lumen could induce significant damage alone or in combination with external sources (Stys et al., 1997).Role of Ryanodine Receptor in White Matter Injury
Thorell et al.(2002) examined the role of intracellular sourced Cain posttraumatic axonal conduction loss following a compressive injury to the spinal cord.They observed that pretreatment with caffeine, a RyR agonist,dose dependently worsened compound action potential recovery after compressive spinal cord injury (SCI), whereas pretreatment with either RyR antagonists’ ryanodine or dantrolene improved compound action potential recovery.
Ouardouz et al.(2003) used an oxygen/glucose deprivation model and showed that the severe propagation failure of dorsal column fibers was surprisingly not recoverable in the absence of extracellular Ca, suggesting that intracellular sources of Ca, and/or Ca-independent processes may also play a role.In support of the former, removal of external Cafrom the perfusate combined with chelation of internal Cain axons conferred robust protection implicating axonal Castores as a source of pathological Ca(Ouardouz et al., 2003).Further manipulations showed that depolarization and activation of Nachannels contributed to conduction block implicating ischemic membrane depolarization, Nainflux, or both processes in mediating Carelease from intra-axonal Castores (Ouardouz et al., 2003).To rule out a role for Nainflux, they substituted Lifor Nain the external bath solution.Li,a monovalent cation, permeates Nachannels and depolarizes axons revealing that ischemic membrane depolarization, rather than Nainflux promoted Carelease in axons (Ouardouz et al., 2003).Importantly, live imaging of Cadynamics directly in axons, in the absence of external Cain the perfusate,established a mechanistic link between ischemia-induced depolarization and mobilization of luminal Cain axonal injury.
The Role of Ryanodine Receptor in Secondary Axonal Degeneration after Spinal Cord Injury
To assess the role of AR Carelease in secondary axonal injury, we developed a two-photon excitation laser-induced SCI model to simultaneously follow dynamic morphological and Cachanges in living myelinated dorsal column fibers acutely after injury (Stirling et al., 2014) (Figure 1).The major advantage of this model from a therapeutic viewpoint is the ability to visually differentiate in real-time primary injured axons (i.e., directly transected) from indirectly injured neighboring axons (i.e., secondary “bystander” injured axons).We found that both “primary” transected axons and “secondary”injured axons both experienced robust increases in axonal Cathat propagated along the length of the axon before undergoing dieback and spheroid formation respectively (Stirling et al., 2014) (Figure 1).Importantly,the removal of external Cafailed to protect axons from secondary degeneration.However, targeting of both AR Carelease receptors (RyR and IPR) was highly protective and reduced secondary “bystander” degeneration of axons (Stirling et al., 2014).Conversely, a RyR2 gain-of-function mutant(RyR2 R4496Cmutant mice) as well as caffeine-induced RyR mediated Carelease worsened these events (Stirling et al., 2014).We next showed that delayed inhibition of RyR alone by ryanodine or RyR knockdown was highly protective and reduced axonal dieback and secondary axonal degeneration after SCI (Orem et al., 2017).Collectively, although all three RyR isoforms have been localized to both uninjured and injured axons, these data provide evidence that RyR2 may play a detrimental role in “bystander” secondary axonal degeneration after SCI (Stirling et al., 2014; Orem et al., 2017; Pelisch et al., 2017).
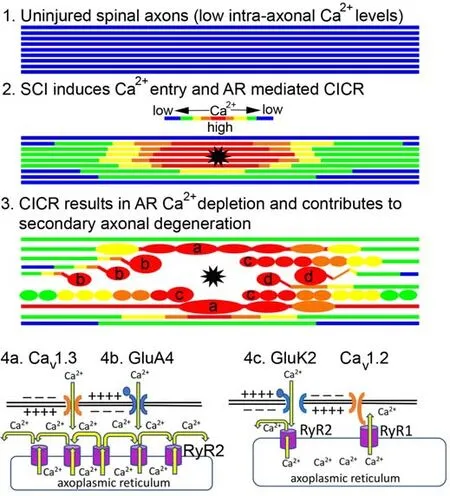
Figure 1|Schematic of proposed RyR-mediated axonal injury pathway.
Putative Roles of Ryanodine Receptor Following Contusive Spinal Cord Injury In Vivo
As the aboveex vivo
studies showed convincing evidence that RyR contributed to axonal injury, we assessed whether RyR inhibition would protect axons after contusive SCIin vivo
.We first assessed the spatiotemporal expression of the three RyR isoforms after a 50 kilodyne, T9/10 contusive SCI from 1 to 7 days after SCI.We found that RyR3 mRNA was significantly increased in lumbar dorsal root ganglion (DRG) and at the lesion site at 1 and 2 days after SCI, whereas RyR2 mRNA was only increased at the lesion site at 1 day after SCI (Pelisch et al., 2017).RyR2 and RyR3 protein expression was localized to lumbar DRG neurons and their spinal projections within the lesion site acutely after SCI (Pelisch et al., 2017).In contrast, RyR1 was unaltered following SCI(Pelisch et al., 2017).These results showed acute differential expression of RyR isoforms in DRG and spinal cord after SCI and revealed that RyR3 in addition to RyR1 and RyR2, may also influence SCI outcome.We then used two-photon excitation intravital microscopy to document the dynamic changes of dorsal column axons following a contusive SCIin vivo
.We found that RyR inhibition within 15 minutes of SCI significantly reduced axonal spheroid formation and prevented secondary degeneration of axons at 24 hours after SCI (Orem et al., 2022).However, delayed inhibition of RyR had no effect on acute axonal spheroid formation but significantly increased axonal survival (Orem et al., 2022).We generated triple transgenicAvil
-Cre:Ai9:Ai95mice that express the genetically-encoded Caindicator GCaMP6f in tdTomato positive DRG neurons that project their axons within the dorsal columns and showed that RyR inhibition reduced intra-axonal Calevels at 24 hours after SCI (Orem et al., 2022).Currently, there is one published study that has assessed the role of RyR in SCI that included assessment of behavioral recovery.Liao et al.(2016)injected lentiviral shRNAi-RyR2 directly into the spinal cord to knock down RyR2 function prior to a moderate contusion injury in the rat.Remarkably,they reported that RyR2 knockdown significantly improved locomotor recovery (Basso, Beattie, and Bresnahan locomotor rating scale and combined behavioral scores) and reduced lesion size by almost 50% compared to controls after SCI (Liao et al., 2016).Given that the contusion was at thoracic level T9, the improved functional outcome likely involved sparing of white matter tracts.The authors then went on to show that mechanistically, the RyR2 knockdown reduced proinflammatory cytokine levels, improved spinal cord oxygen consumption rate, reduced ER stress, and oxidative stress in part through the reduction in NADPH oxidase-2 expression (Liao et al., 2016).Collectively, these studies provide strong evidence that RyR inhibition protects axons and improves functional outcomes after SCI through both direct and indirect effects.
Internodal Axonal Nanocomplexes and Ca2+Dysregulation
To further our understanding of the molecular mechanisms that regulate RyR activation, Stys and colleagues (Ouardouz et al., 2003) revealed a physical association between L-type Cachannels and RyR in white matter.Specifically,Ca1.2 with RyR1 and Cav1.3 with RyR2, and these channels were colocalized within junctions of subaxolemmal AR and axolemma.Ultrastructural studies have also shown AR-axolemmal junctions in axons that resemble the coupling junctions between the sarcoplasmic reticulum and the T-tubules in muscle cells (Metuzals et al., 1997).Thus, analogous to “excitation-contraction coupling” in skeletal muscle, Ca1.2 localized to the axolemma acts as a voltage sensor transducing axonal depolarization to open RyR1 in a Caindependent manner (Ouardouz et al., 2003).In distinction, Cainflux through Ca1.3 or other axolemma localized Capermeable receptors (see below) may trigger CICR through RyR2 like cardiac muscle.Thus, both Ca-dependent and independent pathways could potentially activate AR localized RyR that can release damaging amounts of Caunder pathological conditions in white matter (Ouardouz et al., 2003; Figure 1).
Although excitotoxicity of vital glial elements including oligodendrocytes and astrocytes had previously been implicated in white matter injury (Fern et al.,2014), axons were not thought to express functional glutamate receptors.Remarkably, further work from Stys and colleagues linked axonal localized AMPA receptor activation (GluA4) with RyR-mediated release of Cafrom internal stores (Ouardouz et al., 2006; Figure 1).Thus, excessive glutamate release could directly activate axonal GluA4 receptors that in turn permeate Caleading to CICR through RyR, resulting in pathological Carelease in axons (Ouardouz et al., 2009a).
Further mechanistic work revealed that myelinated spinal axons also expressed functional GluK2 containing kainate receptors that formed axonal signaling “nanocomplexes” along with L-type Cachannels, RyR, and nitric oxide synthase (Ouardouz et al., 2009b).In this arrangement, it was proposed that the axolemma localized GluK2 induce a local depolarization and Cainflux.The depolarization would activate L-type Cachannels and transduce the axonal depolarization to open RyR1 and the Cainflux would promote the release of nitric oxide (NO) by nNOS.NO release in turn may function to increase or modulate RyR activity as has been shown previously in skeletal muscle (Ouardouz et al., 2009b).
Importantly, this binary membrane (i.e., axolemma and AR) localized signaling nanocomplex in axons links glutamate and NO signaling to the pathological release of intra-axonal Cathrough RyR.As traumatic and neuroinflammatory injury processes are known to release abnormal levels of glutamate and NO,this may further perpetuate luminal Carelease and directly contribute to both glial and axonal demise.
Together, the results of several independent studies support a detrimental role for RyR-mediated Carelease in axonal injury, but many questions remain unanswered.Are the positive effects of RyR inhibition after injury a result of direct effects on axons or indirect effects or both? What are the roles of the individual RyR isoforms in white matter injury? Further studies will be necessary to address these questions and how best to target intra-axonal Carelease to protect white matter.
Putative Physiological Roles of Ryanodine Receptor in Myelinated Axons
What are potential physiological roles of RyR in myelinated axons and the internodal nanocomplexes that lead to their activation? Although speculative,the arrangement of a continuous binary membrane signaling nanocomplex in axons and the known existence of a second longitudinal conducting pathway through the internodal periaxonal space (Cohen et al., 2020), may link conduction dynamics anywhere along the length of the axon with an elaborate regenerative intraneuronal signaling system.The latter perhaps through RyR-mediated CICR could provide real-time homeostatic feedback of activity, the local environment, and needs of the axon from any point along the axon to the distant soma.In addition, axomyelinic “synapses” may provide a highly localized link between axonal activity and the resultant metabolic needs of the axon through RyR-dependent signaling facilitating oligodendroglial-derived axonal trophic support (Micu et al., 2017) and/ or through RyR mediated Carelease to axonal mitochondria to increase ATP production.Lastly, a role for RyR-dependent modulation of axomyelinic synapses could underlie activitydependent plasticity resulting in physical changes in myelin that fine tune conduction dynamics (Cullen et al., 2021).More work in these exciting areas of research will likely unveil additional roles for axonal RyR in white matter physiology, plasticity, and injury.
Author contributions:
The author was responsible for all literature searches,data collection, writing and submission of the manuscript, and approved the final version of the manuscript.
Conflicts of interest:
The author declares no conflicts of interest.
Availability of data and materials:
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Open access statement:
This is an open access journal, and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewer:
Pamela J.Lein, University of California Davis, USA.
Additional file:
Open peer review report 1.

- 中国神经再生研究(英文版)的其它文章
- Melatonin, tunneling nanotubes, mesenchymal cells,and tissue regeneration
- Multi-targeted anti-inflammatory drugs for the treatment of neurological disorders
- Emerging roles of GPR109A in regulation of neuroinflammation in neurological diseases and pain
- MicroRNAs as potential biomarkers in temporal lobe epilepsy and mesial temporal lobe epilepsy
- Notice of Retraction
- Roles of constitutively secreted extracellular chaperones in neuronal cell repair and regeneration