
Targeting neuroinflammation in Alzheimer’s disease:from mechanisms to clinical applications
2022-10-22 04:27ZiZhenSiChenJunZouXiMeiXiaoFangLiHuLuoYaoShenJunHuXingXingLiLunWuYuLiu
中国神经再生研究(英文版) 2023年4期
Zi-Zhen Si , Chen-Jun Zou , Xi Mei, Xiao-Fang Li, Hu Luo, Yao Shen, Jun Hu Xing-Xing Li, Lun Wu Yu Liu
Abstract Alzheimer’s disease is characterized by sustained neuroinflammation leading to memory loss and cognitive decline.The past decade has witnessed tremendous efforts in Alzheimer’s disease research; however, no effective treatment is available to prevent disease progression.An increasing body of evidence suggests that neuroinflammation plays an important role in Alzheimer’s disease pathogenesis, alongside the classical pathological hallmarks such as misfolded and aggregated proteins (e.g., amyloid-beta and tau).Firstly, this review summarized the clinical and pathological characteristics of Alzheimer’s disease.Secondly, we outlined key aspects of glial cell-associated inflammation in Alzheimer’s disease pathogenesis and provided the latest evidence on the roles of microglia and astrocytes in Alzheimer’s disease pathology.Then, we revealed the double-edged nature of inflammatory cytokines and inflammasomes in Alzheimer’s disease.In addition, the potential therapeutic roles of innate immunity and neuroinflammation for Alzheimer’s disease were also discussed through these mechanisms.In the final section, the remaining key problems according to the current research status were discussed.
Key Words: Alzheimer’s disease; astrocytes; immune signaling; inflammatory cytokines; microglia;neuroinflammation; neurotoxicity; therapeutic strategies
Introduction
Along with the aging of the global population and recent improvements in medical care, Alzheimer’s disease (AD) has become a burden on the public and healthcare systems worldwide.However, the limited efficacy of currently available treatments has led to an unmet need for more effective therapies.The amyloid cascade hypothesis put forward in the 1990s proposes that misfolded and aggregated amyloid-beta (Aβ) protein triggers the pathological hallmarks of AD, such as neuronal cell death and dementia (Hardy and Allsop, 1991; Hardy and Higgins, 1992).The tau hypothesis put forward in 2013 postulates that tau is a primary factor that promotes the onset of AD(Giacobini and Gold, 2013).Clinical progress based on these two hypotheses has remained slow over the past decades.Ample evidence suggests that neuroinflammation could be a significant driver of AD progression alongside AD’s two classical pathological hallmarks of tau and Aβ aggregation.Investigating the crucial role of neuroinflammation in AD development could help identify novel markers of AD progression and therapeutic targets (Fan et al., 2017; Wang et al., 2017; Zolezzi et al., 2017).There is overwhelming evidence substantiating that in the central nervous system (CNS), microglia and astrocytes are the predominant mediators of inflammation and prime candidates for investigations of neuroinflammatory processes in AD (Hamelin et al., 2016; Schwartz and Deczkowska, 2016; Alves et al., 2017; Daria et al.,2017).The quantification of inflammation levels and inflammatory ligands has revealed that neuroinflammation is one of the earliest detectable indicators of AD (Gispert et al., 2016; Hall et al., 2017).Targeting the physiological processing and inflammation involved in glia, cytokine release,and inflammasome formation is the best approach for cognitive disorders,including AD (Calsolaro and Edison, 2016; Ryan and Kelly, 2016; Toricelli et al.,2021).Growing evidence suggests that glia, cytokines, and inflammasomes are involved in numerous inflammatory diseases, providing the foothold to develop novel treatment strategies (Arancio, 2020; Rafii and Aisen, 2020; Shi et al., 2020; Zhu et al., 2022).Herein, we provide an overview of the roles of the glia-associated inflammation in AD pathogenesis and updated information on the role of microglia and astrocytes in AD pathology.Then, we discuss the dual nature of inflammatory cytokines and inflammasomes that may contribute to AD pathology.In the final section, we provide a brief overview of the therapeutic potential of innate immunity and neuroinflammation for AD, highlighting the beneficial and detrimental roles of inflammation and the feasibility of inflammation as a therapeutic strategy.
Retrieval Strategy
The PubMed database was used to search available literature, using the following combinations of keywords to initially select articles:neuroinflammation and Alzheimer’s disease; immune signaling and neurodegenerative disease; microglia; astrocytes; neurotoxicity; inflammatory cytokine; therapeutic strategies.Most of the selected literature (90% of all references) were published from 2012 to 2022.
Clinical and Pathological Characteristics of Alzheimer’s Disease
AD is a common neurodegenerative disease characterized by memory loss,pronounced cognitive decline, and extensive synaptic and neuronal loss(Sierksma et al., 2020).The neuropathological features for AD patients include intracellular neurofibrillary tangles (NFT) composed of hyperphosphorylated tau protein (p-tau), as well as extracellular amyloid plaques formed by Aβ(Condello et al., 2020; Wen et al., 2022).The production of neurotoxic Aβ depends on the amyloidogenic pathway of amyloid precursor protein (APP)cleavage, driven by β-secretase and γ-secretase (Figure 1).Current evidence suggests that pathological manifestations of Aβ deposition arise earlier than clinical symptom onset.The formation of NFTs leads to the onset of neuronal damage and AD (Arancio, 2020; Franzmeier et al., 2020; Figure 2).However,the pathological progression for Aβ and Tau in different brain regions is heterogeneous.
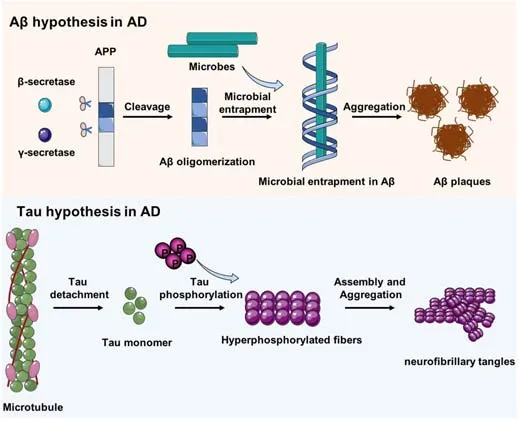
Figure 1| The Aβ and tau hypotheses in AD.
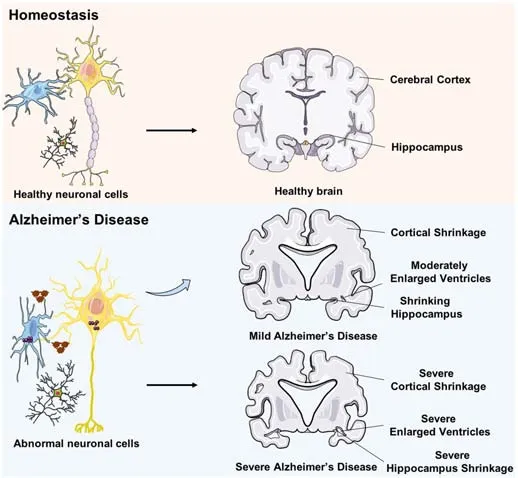
Figure 2 | The difference between healthy brains and those of patients with AD.
Along with these two specific neuropathological hallmarks, AD exhibits alterations in immune responses and neuroinflammation.Quantification of inflammation levels using positron emission tomography and inflammatory ligands revealed that neuroinflammation is also the earliest detectable finding in AD (Gispert et al., 2016; Hall et al., 2017).It is well established that, in the CNS, microglia and astrocytes are the predominant mediators of inflammation(Hamelin et al., 2016; Schwartz and Deczkowska, 2016; Alves et al., 2017;Daria et al., 2017).
Microglia are the resident innate immune cells in the brain, derived from primitive hematopoietic progenitors.By adopting different states in AD,microglia migrate to damage sites, secrete various inflammatory molecules,and phagocytose debris and aggregated proteins (Prinz et al., 2019).It is becoming increasingly clear that microglia do not just play a secondary role in AD processes but virtually contribute to neuron loss, synaptic dysfunction,and the buildup of neurotoxic proteins in the very early stages of AD (Leng and Edison, 2021).
Astrocytes produce neurotransmitters and stimulate synaptogenesis and synaptic neurotransmission, forming part of the blood-brain barrier (Yu et al.,2020).Studies in rodent models suggest a direct contribution of astrocytes to neuroinflammatory processes in AD.Moreover, astrocytes regulate the clearance of Aβ and tau (Price et al., 2021).
The Role of Immune Cells in Alzheimer’s Disease
Microglia
Microglia arise from the yolk sac during development and constitute 0.5-16%of all glia in human brains and 5-12% in rodent brains (Hamelin et al., 2018).Historically, microglia have been classified into different subtypes such as quiescent (also called resting or ramified), activated, and phagocytotic (also called ameboid) cells according to their morphology, electrophysiological properties, density, and the surface expression of immune molecules(Stratoulias et al., 2019).The implementation of single-cell RNA sequencing and cytometry by time-of-flight mass spectrometry has demonstrated distinct gene expression profiles of microglia under different conditions, providing a high-resolution view of the developmental and spatial heterogeneity of microglia across multiple brain regions (Masuda et al., 2020).Microglia are widely acknowledged to be critical for the development of the CNS and contribute to the regulation and connection of neurons and support neuronal development.For example, microglia are responsible for synaptic pruning to eliminate inappropriate synapses and ensure normal neuronal activity.Microglia also regulate learning, memory, and cognitive functions in the adult brain (Udeochu et al., 2018).
As the resident immune cells in the CNS, microglia in the brain mediate the acute immune response to harmful stimuli, such as misfolded Aβ peptides.However, the benefits and physiological functions of acute microglial activation may be abrogated if the acute response associated with harmful stimuli is not resolved (Johansson et al., 2018; Figure 3).This means that the ‘activation’ of microglia has both beneficial and detrimental roles in the pathogenesis of AD (Venegas et al., 2017; Zhao et al., 2018).Acute microglial activation contributes to decreased accumulation of Aβ via increased clearance or phagocytosis (Ziegler-Waldkirch et al., 2018), whereas chronic microglial activation may provoke dysregulated neuroinflammatory responses and lead to neuronal distress, synapse loss, Aβ production, and neurotoxicity via multiple pro-inflammatory cascades (Shi et al., 2019; Ng et al., 2020;Zhang et al., 2020).

Figure 3|The acute response during homeostasis and the chronic response in AD.
Genome-wide association studies have identified frequent variants in AD patients associated with mutations in pattern recognition receptors (PRRs), an evolutionarily conserved family of innate immune cell receptors expressed by microglia, and disease risk.These PRRs, including TREM2, RAGE, sialic acidbinding immunoglobulin-like lectin (Siglec) 3 (also known as CD33), Siglec 2(also known as CD22), dectin-1 (also known as CLEC7A), and the complement receptors C1qR and CR3, enable microglia to sense and respond to various immunostimulatory signals in the brain (Deczkowska et al., 2018; Hopperton et al., 2018).Moreover, PRRs help microglia bind to different species of Aβ in brains with various affinities.The administration of the neurotoxic oligomeric Aβ species activates microglia, decreases ramification, and upregulates microglial CD68 levels in mice brains (Yang et al., 2017).One of the bestcharacterized AD-related PRRs is TREM2 (Ulland and Colonna, 2018).It has been shown that TREM2 recognizes the Aβ chaperone APOE in the AD brain and the lipids and Aβ released from damaged neurons (Guerreiro et al.,2013).
Astrocytes
Astrocytes (also astroglia) are glia that play a critical role in supplying nutrition to neurons; regulating neurotransmission; the modulation of synapse formation, maturation, and elimination; and calcium homeostasis (Habib et al., 2020).Astrocytes exhibit altered morphology in different CNS disorders,which may reflect different functions and accelerate disease pathogenesis(Harris et al., 2020).Induced pluripotent stem cell-derived astrocytes from patients with familial and sporadic AD reportedly exhibit a less complexmorphological appearance and pronounced pathological phenotype than control cells (Jones et al., 2017).
The pathological response of human astrocytes includes reactive astrogliosis and astroglia atrophy (Johnson et al., 2020).These two distinct phenotypes are both observed in the brains of patients with AD (Hsu et al., 2018).Ample evidence suggests that astrocytes are reactivated by microglia and play an essential role in neuroinflammatory and neurodegenerative processes in AD(Liddelow et al., 2017; Rothhammer et al., 2018).These findings revealed the synergistic action of astrocytes and microglia.Accordingly, reactive astrocytes have been documented at sites near activated microglia, around Aβ plaques, and in the neurodegenerative regions of post-mortem AD braintissue.Atrophic astrocytes can be observed in the early stages of AD animal models, which may affect synaptic connectivity because of reduced astrocyte arborization, leading to progressive cognitive deterioration (Yeh et al., 2011;Kulijewicz-Nawrot et al., 2012).Interestingly, several studies have shown that Aβ plaques surrounding astrocytes exhibit a reactive phenotype, even though atrophic astrocytes are not related to senile plaques (Olabarria et al., 2010;Beauquis et al., 2013).
Reactive astrocytes are not only related to neural degeneration but also facilitate inflammation in AD.Higher complement factor content, such as C1q and C3, have been found in exosomes obtained from astrocytes of AD patients, suggesting the possible contribution of astrocytes to AD pathogenesis by secreting complement factors (Goetzl et al., 2018).The reactivity of astrocytes can be initiated and modulated by multiple inflammation-related signaling pathways (Butchi et al., 2010; Schachtrup et al., 2010; Gorina et al., 2011), such as the NF-κB pathway (Akama et al., 1998),MAPK pathway (Agusti et al., 2011), the JAK/STAT3 pathway (Ben Haim et al.,2015a, b), and the calcineurin pathway (Furman and Norris, 2014; Table 1).These results indicate the critical role of astroglial inflammation in AD (Furman et al., 2012).

Table 1 |Astrocyte reactivity modulating signaling pathways
Cytokines and Immune Signaling in Alzheimer’s Disease
Over the years, many natural immune cytokines, such as tumor necrosis factor-α (TNF-α), interleukin (IL)-10, IL-12, IL-18, IL-23, IL-33, IL-34, IL-1β,and type I interferon, have been suggested to be tightly related to diverse AD processes (Mizuno et al., 2011; Bhaskar et al., 2014; Tan et al., 2014;Guillot-Sestier et al., 2015; Fu et al., 2016; Taylor et al., 2018; Thawkar and Kaur, 2019; Melo et al., 2020; Si and Wang, 2020).Elevations in proinflammatory cytokines in the blood and cerebrospinal fluid (CSF) of patients with AD indicate their important roles in AD development.Moreover,Aβ deposition and its resultant neurotoxicity provide ligands to activate microglial Aβ phagocytosis and promote the production of microgliamediated pro-inflammatory cytokines (Lai et al., 2017).This process can significantly promote the clearance of Aβ and help the brain parenchyma rebuild homeostasis (Polazzi and Monti, 2010; Clayton et al., 2017).Under chronic inflammation, these inflammatory mediators lose effectiveness in clearing Aβ and sustain the inflammatory response in a feed-forward manner to propagate neurotoxicity (Yamamoto et al., 2007).Catalytic factors for this detrimental and sustained response include injury, genetics, age,and peripheral inflammation (Gorle and Vandenbroucke, 2019).Moreover,specific pro-inflammatory cytokines (e.g., interferon-γ) have been shown to cause additional Aβ secretion while activating microglia in response to the neurotoxic Aβ (Roy et al., 2020).
TNF-α
TNF-α is well-recognized as a cytokine that is important for immune function and essential for neuronal functions.Glia-derived TNF-α has been shown to regulate homeostatic synaptic scaling (Stellwagen and Malenka, 2006).Interestingly, higher CSF TNF-α levels have been found in individuals with AD,while individuals with mild cognitive impairment and high levels of TNF-α in the CSF are more likely to develop AD (Tarkowski et al., 2003).Other studies have also substantiated that inflammation markers, such as C-reactive protein and IL-6, were increased in serum and CSF even before signs of increased Aβ or tau were present (Schuitemaker et al., 2009).
However, no consensus has been reached on the effects of TNF-α on AD.Certain studies have shown that inhibiting TNF-α signaling has a therapeutic effect (Contreras et al., 2020).For example, TNF receptor 1-deficient APP/PS1 mice exhibited enhanced choroid plexus tissue preservation, decreased levels of Aβ, CSF blood-brain barrier integrity, lower expression of inflammatory factors, and preserved memory compared with TNF receptor 1-sufficient APP/PS1 mice (Steeland et al., 2018).In contrast, other studies have demonstrated beneficial or benign roles of TNF-α (Steeland et al., 2018).Injection of a TNF-α adeno-associated virus resulted in enhanced microglial responses and attenuated Aβ deposition, suggesting that TNF-α may promote plaque clearance under specific spatial and temporal contexts (Chakrabarty et al., 2011).Studies have also found that anti-TNF therapy for arthritis was associated with a lower risk of AD, suggesting its potential for AD treatment(Camargo et al., 2015; Chou et al., 2016).These inconsistencies in TNF-α in AD may be attributed to the fact that these studies investigated different stages of AD.However, the proposed causal relationship between TNF-α and AD remains subject to debate.The risk of developing AD and peripheral TNF-α expression levels showed no causality in the Mendelian randomization modeling of genome-wide association studies (Yarwood et al., 2016).Thus,the precise role of TNF-α in AD warrants further studies.
Interleukin family members
The pro-inflammatory cytokines IL-1β, IL-18, IL-33, and all members of the IL-1 family have been shown to modulate AD pathogenesis (Su et al., 2016;White et al., 2017).In this regard, it has been shown that inhibition of the IL-1 receptor decreases tau phosphorylation, alleviates cognitive deficits,and modestly modulates Aβ levels in 3×TgAD mice (Sutinen et al., 2012).In contrast, Aβ levels were markedly decreased in 3×Tg AD mice overexpressing IL-1β, highlighting that neuro-inflammatory cytokines affect AD (Heppner et al., 2015).Moreover, IL-18 can promote the production of Aβ40 through β-secretase-mediated cleavage of APP, while loss of IL-18 in APP/PS1 mice incites seizures via impaired dendritic pruning and excessive excitatory synapse activity (Tzeng et al., 2018).IL-33 is downregulated in AD brains,indicating that IL-33 functions as a potential modulator for AD (Liew et al.,2016).Moreover, intraperitoneal injection of IL-33 into APP/PS1 AD mice could decrease Aβ levels, improve contextual memory, and restore longterm potentiation deficiency (Chapuis et al., 2009).IL-34 has also been reported to have conflicting roles in AD pathology.It has been shown that intracerebroventricular injection of IL-34 improves memory and decreases oligomeric Aβ levels through heme oxygenase-1 and insulin-degrading enzyme(Gomez-Nicola et al., 2013).However, in primary microglial cultures from human brains with AD, IL-34 stimulation led to the downregulation of CD68,a lysosomal marker commonly used to evaluate phagocytic potential (Walker et al., 2017).Growing evidence has substantiated the role of IL-23, IL-12, and IL-10 in AD pathogenesis.For instance, the shared subunit of IL-23 (i.e., p40),IL-12, and IL-10 is upregulated in the CSF of AD patients.Furthermore, IL-10 and p40 levels have been reported to be elevated in patients with normal cognitive function and abnormally high Aβ deposition levels (Vom Berg et al., 2012).Moreover, p40 ablation in APP/PS1 mice mitigates Aβ levels and cognitive deficits (Taipa et al., 2019).In addition, p40 silencing in the senile senescence-mediated prone-8 mice attenuates the Aβ load, cognitive decline,and neuronal death (Pedrini et al., 2017).Interestingly, inhibition of IL-23 and IL-12 signaling via p40 genetic ablation could only decrease the number of Aβplaques in males (Eede et al., 2020).Besides, IL-10 has been reported to play two parts regarding its biological role in neurodegenerative processes (Porro et al., 2020).Therefore, the uncertain function of these cytokines severely affects brain function and health during chronic inflammation of AD.
TREM2 signaling pathway
TREM2 is a type I transmembrane receptor uniquely expressed in microglia(Zhang et al., 2014).TREM2 expression has long been thought to be decreased by pro-inflammatory stimuli and mediate anti-inflammatory effects, primarily based onin vitro
studies.However, recentin vivo
studies found that TREM2 expression is increased in inflammatory circumstances and can bind different lipids and apolipoproteins (Wang et al., 2015; Gratuze et al.,2018).More importantly, TREM2 is a receptor for Aβ that mediates microglia function (Zhao et al., 2018).The signaling pathway following TREM2 activation relies on its adaptor protein binding partner DAP12.Binding to DAP12 is primarily required for the TREM2 signaling cascade, as TREM2 lacks an intracellular signaling domain.Phosphorylation of immunoreceptor tyrosinebased activation motif in DAP12 recruits spleen tyrosine kinase (Konishi and Kiyama, 2018), which in turn activates PI3K signaling and subsequently activates downstream signaling pathways, including NF-κB, ERK, and AKT (Peng et al., 2010).In 2013, two independent studies published back-to-back found that R47H substitution caused by a rare single nucleotide polymorphism in TREM2 confers a higher risk of AD than any AD risk gene other than APOE (Guerreiro et al., 2013; Jonsson et al., 2013).Later, another R62H substitution in TREM2 was identified (Sims et al., 2017).It has been established that R47H and R62H produce two loss-of-function TREM2 proteins causing low affinity for TREM2 ligands.Furthermore, many other TREM2 variants, such as D87N, L211P,T96K, and H157Y, have been associated with AD (Jin et al., 2014; Ghani et al.,2016; Jiang et al., 2016).These findings prompted researchers to explore the function of TREM2 in AD.
A Dominantly Inherited Alzheimer Network study involving 16 centers worldwide discovered that soluble TREM2 in CSF is produced in AD in a disease progression-dependent manner (Suarez-Calvet et al., 2016).Microglia in TREM2-deficient mice and patients with the TREM2 R47H mutation exhibit enhanced autophagy due to dysregulated mTOR signal transduction(Krasemann et al., 2017).The secretion of soluble TREM2 produced from proteolytic cleavage of TREM2 can ameliorate the progression of AD in mouse models.In addition, supplementation of soluble TREM2 into 5×FAD mouse brains improves Aβ clearance, promotes microglia proliferation, and prevents further neurotic dystrophy (Wang et al., 2015; Piccio et al., 2016).Furthermore, higher soluble TREM2 levels in CSF have been found in patients with AD than in control individuals.Accordingly, levels of soluble TREM2 has been suggested as a possible AD biomarker (Zhong et al., 2019).
TREM2 signaling can also be inhibited by immunoreceptor tyrosinebased inhibition motif (ITIM) (Washington et al., 2002).Siglec 3 is a wellcharacterized AD risk gene that contains a classical ITIM domain.The binding of Siglec 3 to TREM2 has been shown to yield an inhibitory effect on TREM2 signaling (Griciuc et al., 2019).Nevertheless, it must be noted that these observations in mouse models are questionable.For instance, mouse Siglec 3 is not the real homolog of human Siglec 3, as it lacks the ITIM domain.Thus,the TREM2-CD33 interactions in human microglia and how this interaction affects TREM2 signaling and contributes to AD pathogenesis need further investigation.
Inflammasomes
The inflammasome is a cytosolic immune signaling complex that senses receptor activation and subsequently induces inflammation and inflammatory cell death known as pyroptosis (Man et al., 2017; Xue et al., 2019).Inflammasome formation initiates a signaling cascade to active caspase-1,which induces the cleavage of inflammatory pro-IL-18 and pro-IL-1β into their mature forms.Meanwhile, active caspase-1 can cleave Gasdermin-D(GSDMD), which translocates and forms pores on the cell membrane to promote the outflow of pro-inflammatory cytokines and cell lysis (Figure 4;Yin et al., 2018).Different types of inflammasomes have been documented in the literature, including NLRP1b, NLRP3, NOD-like receptor family apoptosis inhibitory protein (NAIP)-NLRC4, absent in melanoma (AIM) 2, caspase-11,and pyrin (Xue et al., 2019).

Figure 4 | The activation of the NLRP3 inflammasome in AD.
Recent studies have provided novel insights into the function of inflammasome complexes in the AD pathological process (Pereira et al.,2019).For example, in AD, NLRP3 inflammasomes are activated, and their expression increases.Activated NLRP3 inflammasomes promotes the aggregation and hyperphosphorylation of tau proteins (Saresella et al., 2016;Ising et al., 2019; Stancu et al., 2019).Furthermore, overwhelming evidence suggests that ablation of NLRP3 inflammasomes decreases Aβ deposition and attenuates long-term potentiation and spatial memory deficits in APP/PS1 mice (Mitroulis et al., 2010; Goldmann et al., 2013; Pennisi et al., 2017;Voet et al., 2018).In addition, it has been shown that the adaptor protein apoptosis-associated speck-like protein containing a CARD (ASC) can bind with Aβ, and this interaction initiates the activation of NLRP3, leading to the prevention of Aβ clearance (Venegas et al., 2017).This phenomenon leads to a feed-forward vicious cycle.Interestingly, exposure to ASC-Aβ composites promotes microglial pyroptosis.Pyroptotic microglia release ASC that can be functionally built into the neighboring microglia NLRP3 inflammasome (Friker et al., 2020).Accordingly, inhibitors of the NLRP3 inflammasome, such as JC-124 and the fenamate non-steroidal anti-inflammatory drugs mirror the progress made in AD treatment (Daniels et al., 2016).Further, AIM2 loss-offunction protein can reduce Aβ deposition in the brains of 5×FAD mice (Wu et al., 2017) but is not able to sufficiently restore memory capability, as the loss of AIM2 promotes IL-6 and IL-18 expression and preserves IL-1 levels in the brain and, thus, constitutes a compensatory effect to induction of Aβ deposition.
Neuroinflammation-Targeting Therapeutic Strategies for Alzheimer’s Disease
Although the United States Food and Drug Administration has recently approved aducanumab for the treatment of AD, we cannot ignore the fact that many multinational pharmaceutical companies and research institutes spend billions of dollars on anti-Aβ or anti-tau strategies that fail to obtain results, emphasizing the need for novel approaches.
The hypothesis that microglia activation promotes AD progression has prompted researchers to develop microglia depletion strategies using pharmacology or genetics.Microglia depletion in AD mouse models has been shown to have beneficial effects.CSF1R is well-established as a critical surface receptor for microglia (Elmore et al., 2014).CSF1R inhibitors can suppress neuritic plaque accumulation, dendritic spine loss, neuroinflammation, and improve cognition in different AD mice models (Dagher et al., 2015; Sosna et al., 2018).
Furthermore, improving certain aspects of microglial function represent novel therapeutic strategies for AD treatment, targeting innate immune signaling and immunometabolism in microglia.TREM2-activating antibodies, including antibody 1 and antibody 2 identified by Amgen and AL002 and AL002a/c identified by Alector, can reportedly boost myeloid cell function, abrogate survival defects, and can potentially be therapeutic in AD (Cheng et al., 2018;Price et al., 2020; Wang et al., 2020).In an AD mouse model, only AL002a increased microglia aggregation around amyloid plaques, thus improving cognition in 5×FAD mice.Another antibody clone, 4D9, has been reported to be a very potent binder to the extracellular domain of TREM2 (Schlepckow et al., 2020), causing selective reduction of soluble oligomers in an APP knock-in mouse model.
Chemical inhibition of the inflammasome pathway is also potentially effective.Pterostilbene is a natural compound that attenuates the neuroinflammatory response induced by Aβ by inhibiting the NLRP3/caspase-1 inflammasome pathway in microglia (Li et al., 2018).Sulforaphane is an active isothiocyanate component obtained from broccoli seeds and sprouts and exerts an antiinflammatory effect on human microglia by inhibiting NLRP3 inflammasome activation and IL-1β production (An et al., 2016).Moreover, synthesized docosahexaenoic-acid-acylated astaxanthin diesters alleviate cognitive decline by reducing inflammasome formation in APP/PS1 mice (Che et al.,2018).Artemisinin also exhibits inhibitory effects on neuroinflammation and amyloidogenesis by inhibiting NLRP3 inflammasome activation as well as NF-κB production (Shi et al., 2013).Interestingly, β-hydroxybutyrate, one of the main components of ketone bodies, can reportedly reduce reactive oxygen species secretion among patients with AD through activation of Nrf2 signaling, along with the deactivation of the NLRP3 inflammasome and NF-κB to decrease neuroinflammation and thus improves memory decline (Pinto et al., 2018).
The P2X7 receptor (P2X7R) is another regulator for amyloid-mediated NLRP3 inflammasome activation.P2X7R is critical for the Aβ peptide-mediatedrelease of the chemokine ligand CCL3, associated with pathogenic CD8T cell recruitment in AD animal models (Martin et al., 2019).Moreover, Aβ can induce accumulation of IL-1β in microglia from wild-type but not from P2X7-deleted mice (Sanz et al., 2009).The activation of P2X7R can also boost the release of IL-6, CCL2, and TNF-α in microglia (Shieh et al., 2014).Therefore, antagonizing P2X7 signaling is a promising approach for inhibiting neuroinflammation in AD patients.Interestingly, it has been shown that brilliant blue G, a highly selective and blood-brain barrier-permeable P2X7R antagonist, blocks Aβinduced inflammatory responses and diminishes spatial memory impairment and cognitive deficits in a mouse model (Chen et al., 2014).Another selective P2X7R antagonist, A740003, can block ATP-dependent IL-1β release (Facci et al., 2018).Many chemicals have been developed to antagonize P2X7 signaling in the brain in recent years.However,only a few chemicals can cross the blood-brain barrier.Thus, pharmacokinetic properties need to be improved to ensure these chemicals are excellent candidates in clinical trials for AD treatment.Certain nutrients or food groups(e.g., vitamin D, vegetables, and fruits) can improve cognitive function.The Multicultural Healthy Diet (NCT03240406) should help formulate specific guidelines based on foods with anti-inflammatory properties for the prevention of cognitive impairment (Scarmeas et al., 2018).
Limitations
This review is subject to several limitations.The primary limitation to the generalization of the review is that the PubMed database was used to search the literature, and incomplete retrieval of identified research was inevitable.Secondly, the researchers behind the cited works come from diverse cultural backgrounds with naturally varying personal opinions regarding specific phenomena, which may affect the rationality of the research.
Conclusion
The critical roles of innate immunity and neuroinflammation in regulating the progression and pathology of AD opens up exciting new avenues for research into the therapeutic strategies for neurodegenerative disorders(Ennerfelt and Lukens, 2020).Increasing evidence suggests that glia,inflammatory cytokines, and inflammasomes are inextricably linked to a healthy neuronal environment.The activation of microglia and astrocytes,including the secreted cytokines and immune signaling pathways, contribute to pathological neuroinflammation responses, leading to neurotoxicity in AD.Targeting neuroinflammation has strong prospects for clinical application for AD treatment.Numerous studies have focused on Aβ generation and processing, but the lack of solid clinical results has forced us to look at other factors (Brosseron et al., 2020; Nugent et al., 2020).The new avenues on microglia, astrocytes, inflammatory cytokines, and inflammasomes have generated a burgeoning field in AD treatment research (Dourlen et al., 2019).Much-needed novel drugs to combat this devastating neurodegenerative disease can be identified by exploring the roles of innate immunity and neuroinflammation.Recent advances in neuroimmunology research and AD have begun to uncover the significant roles of neuroinflammation in the onset, progression, and pathology of AD (Liu and Aguzzi, 2019).Importantly,recent studies have shown that different factors determine the efficacy of AD treatment, including lifestyle, environmental factors, and immune responses,which can be harnessed to develop individualized treatments.
Author contributions:
ZZS and CJZ prepared the manuscript.XM, XFL, JH,XXL and LW searched the literature.HL and YS edited the manuscript.YL contributed to the definition of intellectual content and manuscript review.All authors approved the final version of the paper.
Conflicts of interest:
The authors have no competing interests regarding the publication of this paper.
Open access statement:
This is an open access journal, and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.

- 中国神经再生研究(英文版)的其它文章
- Melatonin, tunneling nanotubes, mesenchymal cells,and tissue regeneration
- Multi-targeted anti-inflammatory drugs for the treatment of neurological disorders
- Emerging roles of GPR109A in regulation of neuroinflammation in neurological diseases and pain
- Potential physiological and pathological roles for axonal ryanodine receptors
- Notice of Retraction
- Roles of constitutively secreted extracellular chaperones in neuronal cell repair and regeneration