
磷掺杂与MoS2光沉积共同促进CdS光催化产氢
2022-10-19 07:43:38刘俊琛黄浩然葛春玉王红强方岳平
广西师范大学学报(自然科学版) 2022年5期
刘俊琛, 黄浩然, 葛春玉, 王红强, 方岳平*
(1.华南农业大学 材料与能源学院, 广东 广州 510642;2.广西低碳能源材料重点实验室(广西师范大学), 广西 桂林 541004)
随着时代变迁与工业的发展,能源消耗不断提高,以石油为首的化石燃料被用于各种领域。化石燃料燃烧时产生大量污染物,太阳能作为一种清洁且取之不尽的能源受到了广泛关注。通过光催化的方法将太阳能转化为氢能是一种利用太阳能的理想手段。光催化剂最初由Fujishima等[1]在1970年代发现。从最早期的TiO2通过光电化学法分解水开始,越来越多的光催化材料被制备出来,例如g-C3N4[2-3]、SrTiO3[4]、WO3[5]、BiVO4[6-7]、ZnO[8]、ZnS[9]、CdS[10-11]等。在众多的半导体光催化剂中,由于CdS的禁带宽度约为2.40 eV,可以被可见光所激发,是一种非常具有潜力的光催化材料[10]。然而,CdS有在光照下自分解与光生电荷快速复合的缺陷。
为了改善以上缺陷,常用的方法有元素掺杂[12-14]、负载助催化剂[15]、构建异质结[16]等。通常来说,在负载了合适的助催化剂之后,光催化剂光生电子或空穴会快速地迁移到助催化剂中进行催化反应,从而使光生电荷的分离率大幅提高。不仅如此,引入合适的助催化剂还会降低光催化剂表面发生氧化还原反应所需的能量屏障[17]。稀有金属如铂[18]、金[19]因为有着较好的助催化活性、导电性和稳定性,已被广泛用于制备高效的光催化体系中。由于稀有金属的产量低、成本高昂,为了低成本地构建可以大量推广的光催化体系,不含稀有金属的助催化剂,如过渡金属氧化物、过渡金属硫化物、氮化物、磷化物、碳化物等,正受到广泛的研究与关注。目前,CoxOy[20]、MoS2[21-22]、CoSx[23]、MoN[24]、NixP[25]、CoxP[26]、MoP[27]、Co3C[28]、Ni3C[29]等非贵金属助催化剂已被用于进行CdS的改性并取得了一定的进展。其中,MoS2具有优异的氢气释放反应活性和较高比表面的2D结构,是一种非常具有前景的助催化剂。目前已有大量关于CdS/MoS2的研究,可以证明负载了MoS2的CdS的电荷分离率有所提高。然而,CdS/MoS2体系在水溶液中进行高速率光催化产氢需要在Na2S、Na2SO3、乳酸等牺牲剂存在的条件下进行,在纯水中的产氢性能并不理想。为了实现在纯水中进行较高速率的光催化反应,构建一种更高效的CdS与MoS2的结合方式是有必要的。CdS已具在可见光下进行光催化的能力,而通过元素掺杂的方法,可以更进一步拓宽其吸光范围,并且抑制光生电子与空穴的复合。
P掺杂策略早已被用于光催化剂的改性中[30],P在CdS中进行梯度掺杂后,由掺杂所产生的缺陷会捕获光生电子,并且会在催化剂内部建立内建电场,从而阻碍光生电子与空穴的复合,大幅提高光生电子与空穴参与光催化反应的效率[31-32]。P的掺杂还可以减小H2吸附的自由能,并且拓宽光催化半导体的光响应范围[31-32]。基于上述策略,磷元素被选用于对CdS进行元素掺杂,再通过原位光沉积的方法在光催化剂表面负载MoS2,目的是制备可以在纯水中产氢的高效光催化体系。
本文以四水合硝酸镉为原料,通过水热法制得CdS纳米棒,再通过磷掺杂和原位光沉积得到光催化剂CdS/MoS2,并对其进行表征和光解水产氢实验,以期为制备高效光催化水解产氢催化剂提供理论指导。
1 实验部分
1.1 实验试剂、材料和仪器
次磷酸钠、95%乙醇、四硫代钼酸铵、四水合硝酸镉、硫脲、硫酸钠(分析纯,阿拉丁试剂(上海)有限公司);5% Nafion 117 溶液(阿拉丁试剂(上海)有限公司);乙二胺(分析纯,国药集团化学试剂有限公司);FTO 导电玻璃(方块电阻:7 Ω,武汉晶格太阳能科技有限公司)。
EL104电子分析天平(梅特勒-托利多仪器有限公司);Ultima IV X射线衍射仪(日本理学);Talos F200S场发射透射电子显微镜(美国 FEI);VG ESCALAB 250 X射线光电子能谱仪(美国 Thermo VG Scientific);CHI-650E电化学工作站(上海辰华仪器有限公司);UV-2550紫外可见分光光度计、RF-5301PC荧光光度计(日本岛津);FLS980荧光寿命光谱仪(英国 Edinburgh Instruments);PLS-SXE300氙灯光源(北京泊菲莱科技有限公司);GC-7900气相色谱仪(上海天美科学仪器有限公司)。
1.2 CdS纳米棒的制备
向容积为100 mL的水热反应釜中加入4.627 g四水合硝酸镉、3.425 g硫脲、65 mL乙二胺,电磁搅拌30 min后,密封,加热到160 ℃,保温48 h。待水热反应釜自然冷却到室温后,将反应所得产物离心,并用体积分数95%的乙醇进行3次洗涤,60 ℃干燥24 h,得到的产物为CdS纳米棒。
1.3 CdS纳米棒的磷化
取2 g 次磷酸钠颗粒,在玛瑙研钵中研磨成白色粉末,向玛瑙研钵中加入0.5 g CdS纳米棒,研磨至混合均匀得到黄色粉末,倒入瓷方舟,放入高温管式炉,以2 ℃/min的速率加热到300 ℃,保温2 h后自然冷却到室温,将产物与200 mL水混合,超声处理30 min,将所得悬浊液离心,用去离子水洗涤3次,60 ℃干燥24 h,得到掺磷CdS纳米棒 (CdS/P) 。
1.4 CdS/P/MoS2的制备
CdS/P/MoS2通过原位光沉积法制备。50 mg CdS/P与100 mL去离子水混合并超声20 min,得到均匀分散的CdS/P悬浊液,加入一定量的四硫代钼酸铵溶液,连续通入N2并充分搅拌30 min,除去悬浊液中的氧气。通气完毕后,向悬浊液连续照射1 h可见光(安装了400 nm截止滤光片的300 W氙灯,输入电压为220 V,电流为20 A)。光照完毕后,抽滤回收沉淀,用去离子水洗涤沉淀3次,再用体积分数95%的乙醇洗涤1次,将沉淀放入60 ℃烘箱中干燥12 h得到CdS/P/MoS2。将CdS/P换成CdS重复上述步骤,制得CdS/MoS2。
1.5 光催化产氢测试
取上述制备的CdS/P/MoS210 mg,超声分散在100 mL去离子水中,密封且持续通入N230 min,除去悬浊液中的溶解氧。除氧完毕的悬浊液用氙灯照射(安装了400 nm截止滤光片, 输入电压为220 V,电流为20 A)后,生成的氢气通过安装了TCD检测器的Techcomp GC 7900进行检测。
1.6 光电化学测试
将10 mg光催化剂、2 mL无水乙醇、100 μL 5%的Nafion溶液混合后超声30 min,得到混合均匀的悬浊液。将400 μL的悬浊液滴在FTO玻璃基底上 (2 cm×6 cm)。使用红外灯将滴有悬浊液的FTO玻璃基底烘干后,将FTO玻璃基底放入瓷方舟内,于管式炉中,氮气保护气氛下,3 ℃/min升温到150 ℃并保温1 h,自然冷却到室温。将FTO玻璃基底切割成1 cm×2 cm,即得光电极。以上述所制备的光电极为工作电极,铂片电极为对电极,Ag/AgCl为参比电极,使用CHI 650E电化学工作站,组成三电极测试系统进行光电化学测试,300 W氙灯作为光源。电化学氢气析出反应的极化曲线测试、瞬态光电流响应测试、莫特-肖基特测试与电化学阻抗测试在0.5 mol/L的硫酸钠溶液中进行,电化学氧气析出反应的极化曲线测试在1 mol/L的KOH溶液中进行。使用0.4 V的外加电压进行瞬态光电流响应测试,极化曲线测试的扫描速度为0.005 V/s,电化学阻抗测试的振幅为0.005 V,频率为0.01~105Hz。莫特-肖基特测试的振幅为0.005 V,频率为1 000 Hz,平带电位通过莫特-肖基特曲线进行估算,相对于Ag/AgCl电极的电势通过以下公式换算
2 结果与讨论
2.1 成分与形貌分析
通过原位光沉积法在CdS/P表面负载MoS2的反应为
CdS→e-+h+,
[MoS4]2-+2e-→MoS2+2S2-。
CdS/P被光激发产生光生电子与空穴,吸附在CdS/P表面的[MoS4]2-被转移到催化剂表面的光生电子还原成MoS2,从而形成CdS/P/MoS2,光生空穴则消耗在S2-的氧化反应中。通过XRD对CdS、CdS/P、CdS/P/MoS2的晶体结构进行解析,如图1所示,CdS、CdS/P、CdS/P/MoS2的衍射峰与(PDF#:65-3414)相吻合。与CdS的衍射峰相比,CdS/P与CdS/P/MoS2的峰向小角度偏移,意味着CdS/P、CdS/P/MoS2有比CdS更大的晶格参数,这是P掺杂的结果;CdS/P/MoS2的衍射峰没有观察到属于MoS2的衍射峰,这是由于MoS2的含量低。
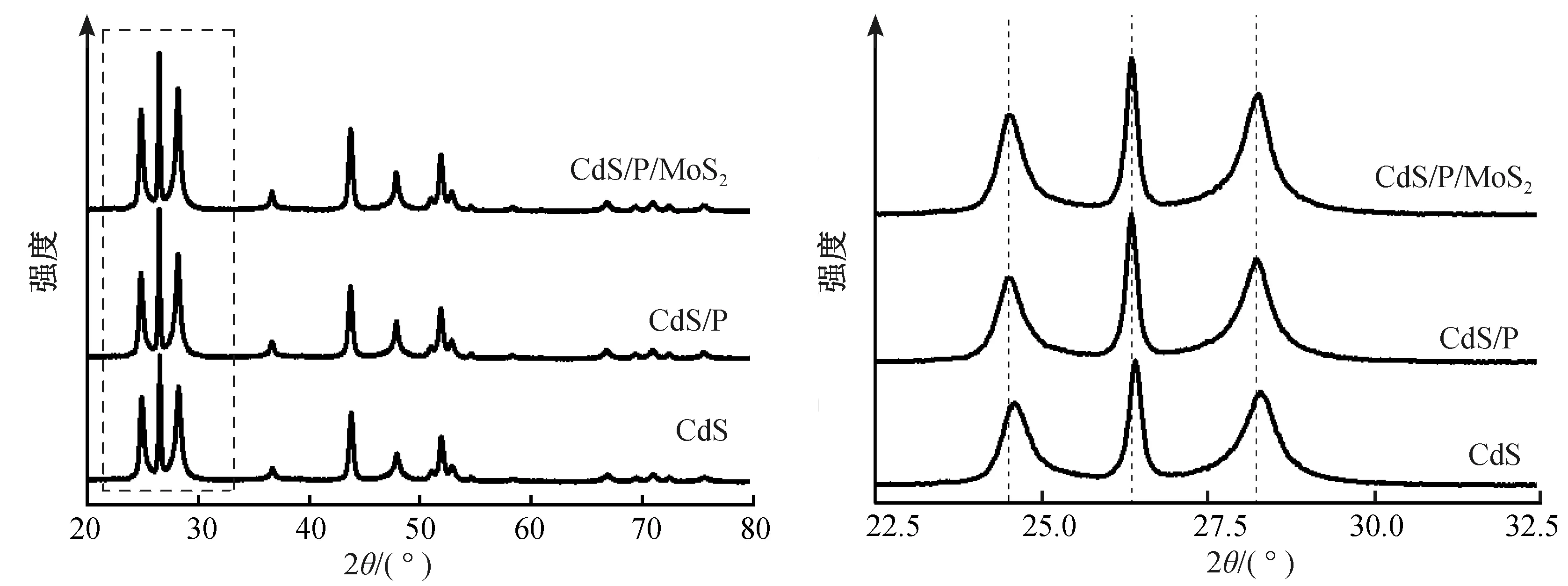
图1 CdS、CdS/P、CdS/P/MoS2的XRDFig.1 XRD patterns of CdS, CdS/P and CdS/P/MoS2
CdS/P/MoS2的形态可以通过TEM进行观察,从图2中可以看出,制备出的CdS/P/MoS2的形态为纳米棒,覆盖在CdS/P/MoS2表面的MoS2可以通过(高分辨透射电子显微镜)HRTEM进行观察。从图3中可以看出,制备出来的CdS/P/MoS2样品表面附着少量的MoS2纳米片,纳米片的晶格条纹宽度为0.303 nm,代表着MoS2的(006)晶面。
CdS/P/MoS2的表面组分通过XPS进行进一步分析。图4为CdS、CdS/P、CdS/P/MoS2的高分辨XPS光谱,其中包括Cd、S、P、Mo元素。从图4(a)中观察得出,CdS中位于411.12和404.36 eV的峰对应着Cd 3d3/2和Cd 3d5/2,证明Cd以Cd2+的状态存在。从图4(b)中可以得出,CdS中位于161.92与160.75 eV的峰对应着S 2p1/2和S 2p3/2,证明S以S2-的状态存在。当CdS进行磷掺杂后,Cd与S的结合能几乎没有改变,由此可得磷掺杂对CdS的化学结构没有明显影响。图4(c)中,CdS-P位于130.07与129.30 eV的峰证明部分P以零价态的形式存在,133 eV处的峰代表着P 2p的峰。上述现象说明在CdS-P中P被间隙掺杂在CdS框架中[31-32]。负载了MoS2以后,CdS-P-MoS2中Cd、S与P的峰明显地向高结合能方向移动,这表明CdS-P与MoS2之间形成了异质结并有利于电荷从CdS-P到MoS2的传导[33]。如图4(d)所示,在233.45与230.45 eV处存在2个微小的峰,分别对应Mo 3d3/2与Mo 3d5/2,说明Mo元素以Mo4+的形式存在[33]。从XPS的结果可以进一步证明MoS2在CdS-P-MoS2中的存在。

图2 CdS/P/MoS2的TEMFig.2 TEM image of CdS/P/MoS2

图3 CdS/P/MoS2的HRTEMFig.3 HRTEM image of CdS/P/MoS2
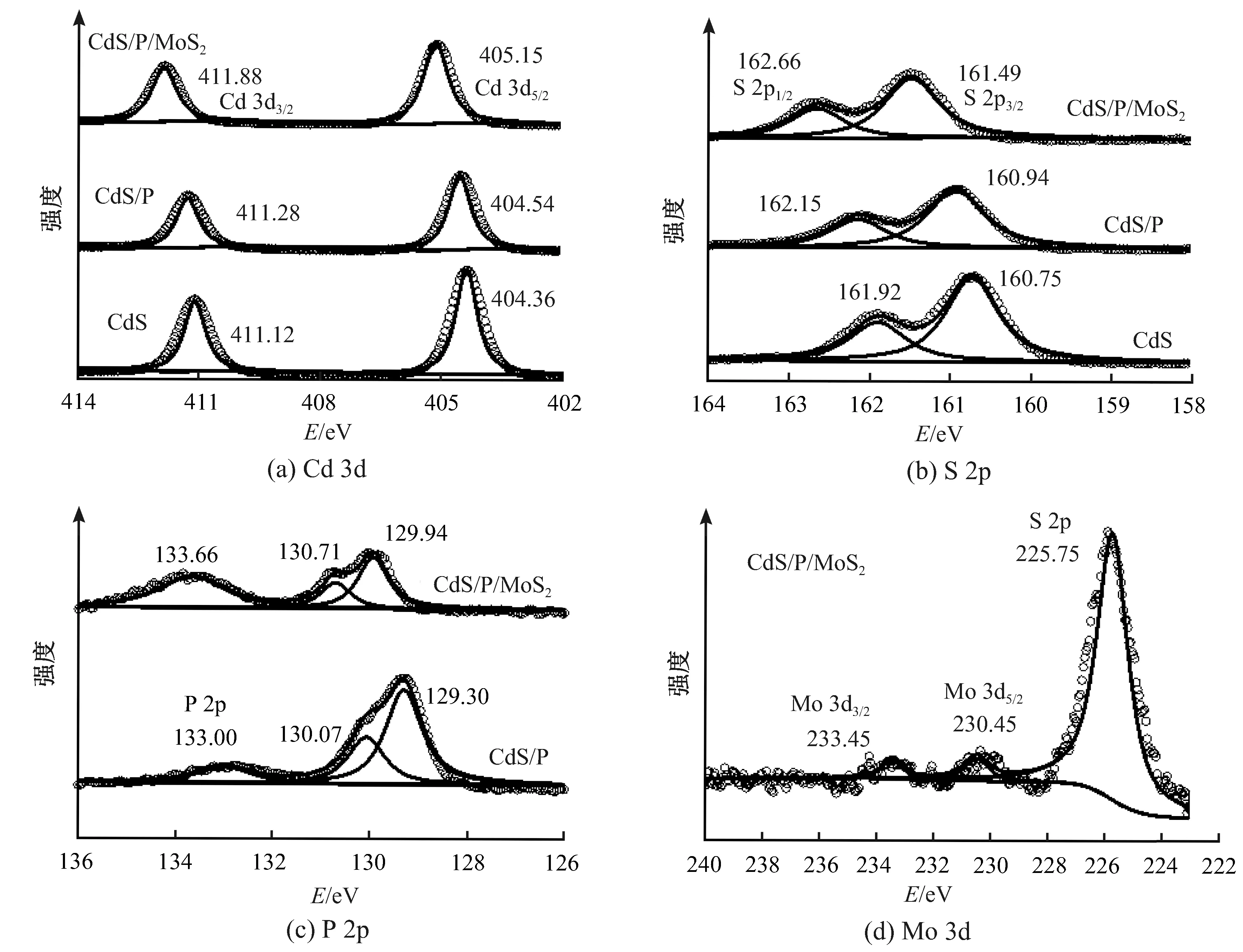
图4 CdS、CdS/P和CdS/P/MoS2的高分辨XPS光谱Fig.4 XPS patterns of CdS,CdS/P and CdS/P/MoS2
2.2 光催化产氢性能分析
图5为制备所得样品的光催化产氢性能图。图5(a)为CdS/P、CdS/MoS2和CdS/P/MoS2的产氢性能对比图,从图中可以看出,CdS单独掺P或单独负载MoS2之后的产氢性能并不理想。CdS/P/MoS2的产氢性能相比CdS/P与CdS/MoS2有大幅提高,说明将P掺杂与MoS2负载相结合可以大幅改进CdS的光催化性能。值得注意的是未改进的CdS样品在纯水中没有光催化产氢能力。为了优化P掺杂与MoS2负载的最佳比例,在制备CdS/P/MoS2时改变CdS与NaH2PO2的比例,或改变四硫代钼酸铵的添加量以改变Mo的理论负载量。如图5(b)、5(c)所示,当CdS与NaH2PO2的比例为1∶4,四硫代钼酸铵的理论负载量为1%时,CdS/P/MoS2的光催化产氢的性能最好,产氢速率为692.9 μmol/(g·h),在420 nm处的表观量子效率为0.31%。图6为CdS/P/MoS2在纯水中进行连续8 h的产氢测试,从图中可以看出样品的性能在8 h内无明显衰减。
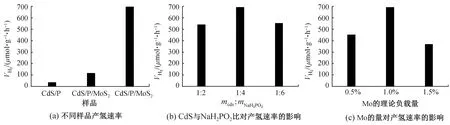
图5 样品的产氢速率Fig.5 Average rate of H2 evolution of samples
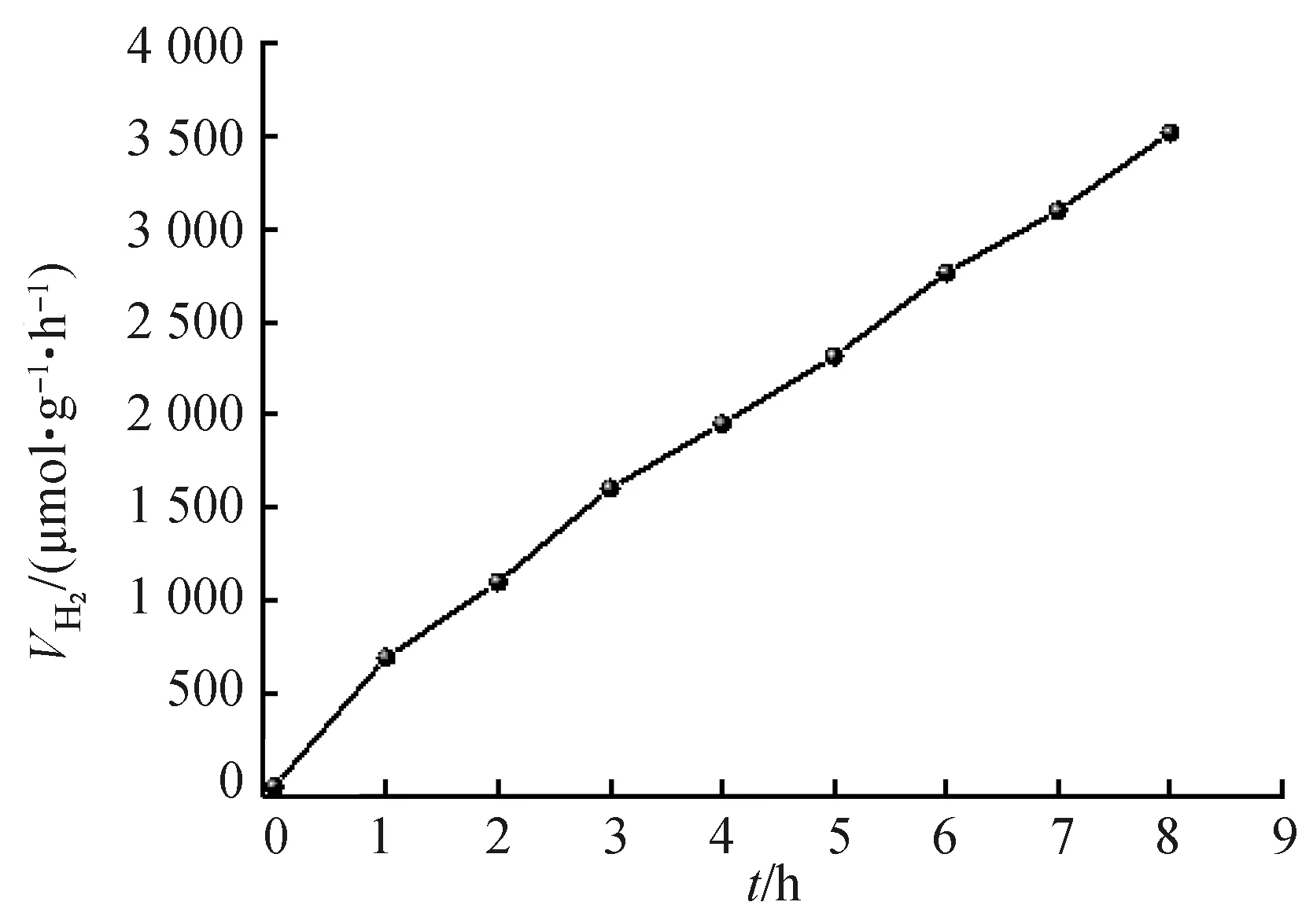
图6 CdS/P/MoS2的产氢稳定性Fig.6 Stability test of CdS/P/MoS2
2.3 光催化产氢机理分析
将光催化剂的吸光范围扩大可以增加其产生的光生电荷。图7为CdS、CdS/P、CdS/P/MoS2的紫外可见吸收光谱图,从图中可以看出,CdS的光吸收带从紫外区延伸到约530 nm。掺磷后,CdS/P的光吸收范围有明显增加,证明P的引入增强了催化剂的光吸收能力。从吸收光谱中发现,当负载了MoS2后,CdS/P/MoS2的光吸收从波长300 nm到800 nm都有明显的增强,这归因于MoS2对光的吸收[34]。CdS、CdS/P、CdS/P/MoS2的带隙可通过Kubelka-Munk法对吸收光谱的计算得到[34]。如图8所示,带隙的数值为切线与(αhv)2=0交点的hv值,其中hv是光子的能量,α是吸收系数。从图中得出,CdS的带隙为2.38 eV,CdS/P的带隙为2.35 eV,CdS/P/MoS2的带隙为2.33 eV。显然P的掺杂与MoS2的负载都有利于提高CdS/P/MoS2对光的吸收能力,如此便能产生更多的光生电荷用于光解水。

图7 样品的紫外光谱Fig.7 UV-vis DRS of samples

图8 样品的紫外光谱换算Fig.8 Corresponding plots of DRS
为了在光催化剂的表面进行氧化还原反应,将光生电子与空穴转移到催化剂表面并阻止其复合是至关重要的。界面电荷转移非常有利于光生电子与空穴向表面迁移与分离。莫特-肖特基测试结果可以确认能带的位置,并帮助解析CdS/P/MoS2中电荷迁移的路径。图9为CdS、CdS/P、CdS/P/MoS2的莫特-肖特基测试结果图,从图中可以发现,CdS、CdS/P、CdS/P/MoS2的曲线斜率为正数,证明这3种样品具有n型半导体的特性[35]。MS曲线中线性部分的切线在X轴上的截距数值即为该样品的平带电位(Vfb)。从图中可得CdS的平带电位为-0.52 V(vs RHE),CdS/P的平带电位为-0.51 V(vs RHE),CdS/P/MoS2的平带电位为-0.32 V(vs RHE)。对比3个样品的平带电位可以发现,CdS/P的平带电位比CdS的更正,这意味着P的掺杂导致CdS/P中的导带底的下移。CdS/P/MoS2的导带底与CdS/P相比向正向移动,这是由于MoS2的费米能级比CdS/P的更正。当CdS/P与MoS2之间形成紧密接触后,CdS/P的电子转移向MoS2导致能带弯曲,使得费米能级达到一个新的平衡[36]。由于CdS/P中的电子向MoS2迁移,导致CdS/P的导带向正电位方向弯曲,而MoS2的导带向负电位弯曲,至此产生了肖特基结,肖特基结将诱导光生电子从CdS/P向MoS2迁移。通过以上所得的平带电位数据和带隙宽度,结合n型半导体的导带比平带更负约-0.1 V的规律,可以得到如图10的能带结构图[37]。从图10中可以得出,光生电子倾向于向MoS2转移,光生空穴倾向于向CdS/P转移。这表明所制备的CdS/P/MoS2光催化体系有利于促进电荷分离,进而提高光催化效率。
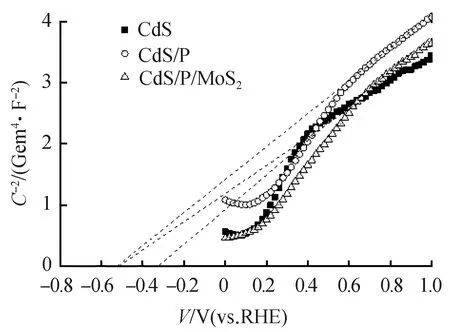
图9 样品的MSFig.9 MS plots of samples
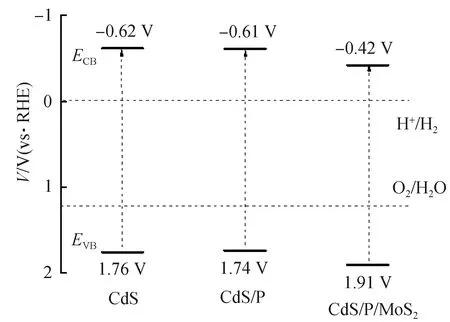
图10 样品的能带示意Fig.10 Schematic band structure diagrams
为了对CdS/P/MoS2的电荷转移和分离能力进行进一步的表征,对其进行光学和光电化学测试。光致发光(PL)光谱可用于测试样品的光生载流子的分离与复合倾向[38]。从图11中可以看出,CdS在540 nm周围有一个较大的荧光发射峰,而CdS/P在相同区域的的荧光强度低于CdS。这意味着CdS中的光生电荷受单一CdS的结构限制较倾向于复合并发出荧光,而CdS/P中的光生电荷因P的掺杂导致能带结构变化,倾向于分离而减少电荷的复合发光。值得注意的是,CdS/P/MoS2的荧光强度比CdS/P更进一步降低。这可以证明MoS2负载后,由于莫特-肖特基能垒的存在,光生电子与空穴被进一步分离,更难以复合。这种结果说明CdS/P/MoS2比CdS和CdS/P具有更高的光利用率,从而提高了光催化的效率。
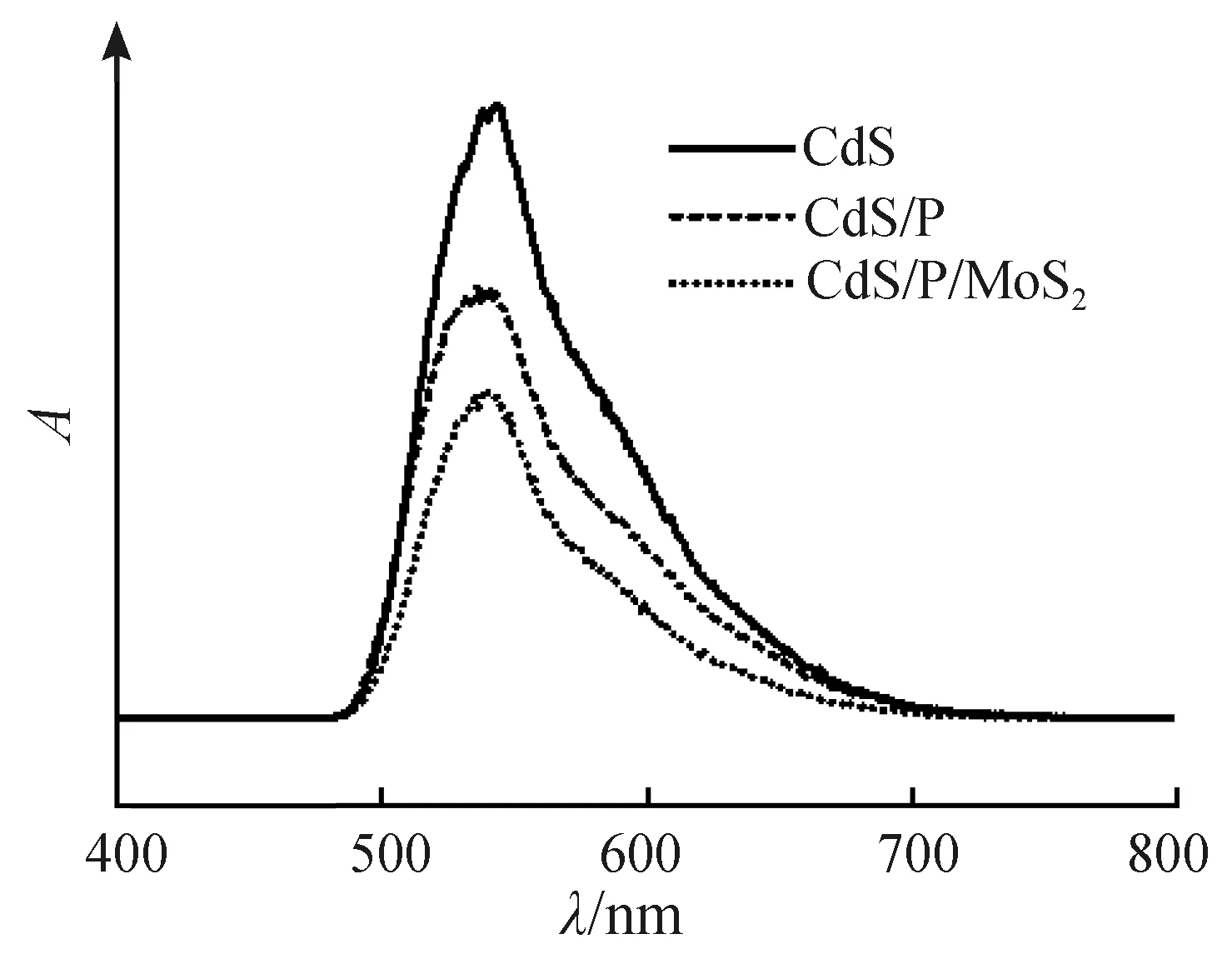
图11 样品的荧光光谱Fig.11 PL spectra
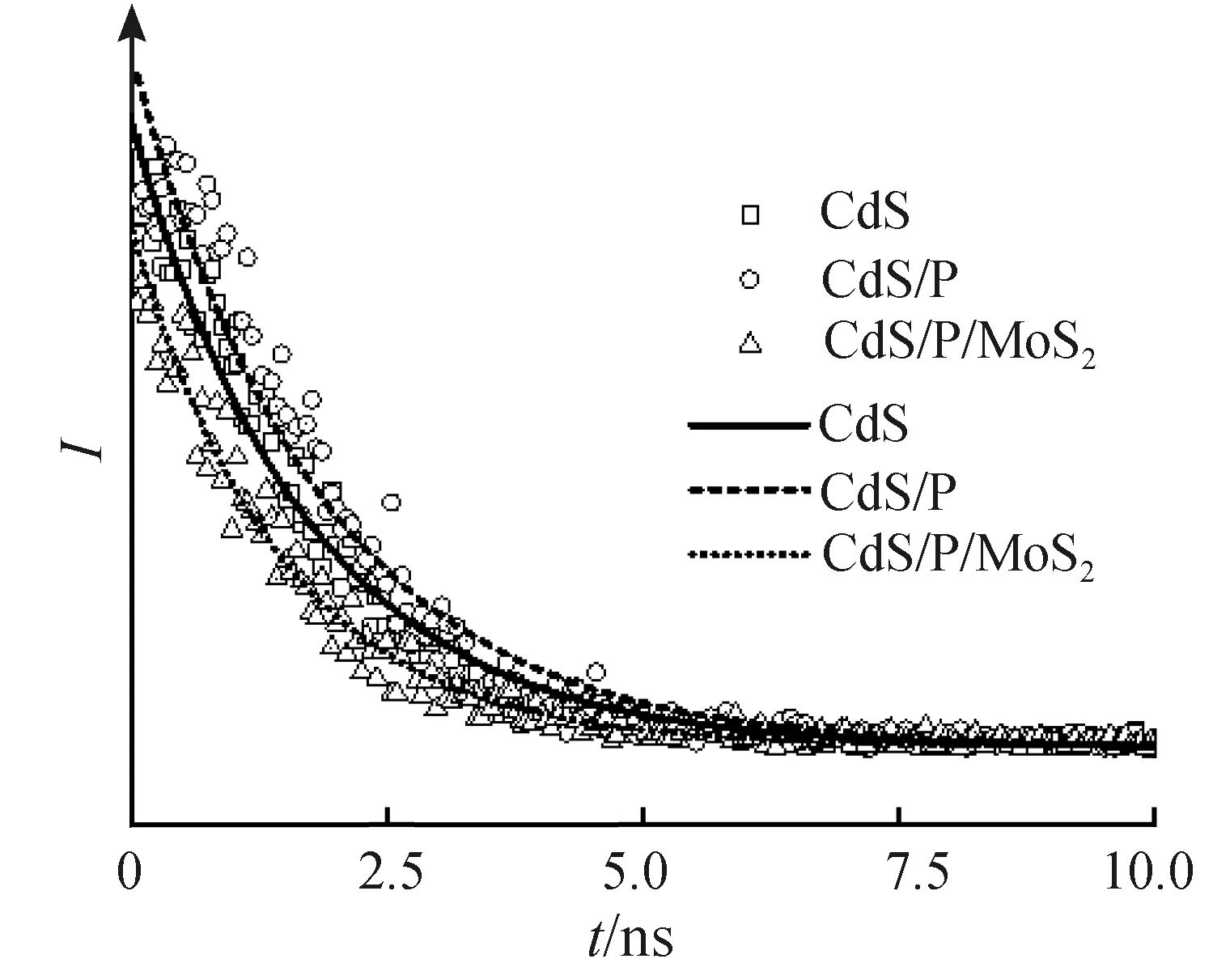
图12 样品的荧光寿命Fig.12 Transient PL spectra
荧光寿命测试可以对样品的光生电荷进行更详细地分析[38]。通过双指数函数拟合(图12)与计算,得到样品的平均荧光寿命见表1。在没有助催化剂的单一光催化体系中,越长的荧光寿命意味着光生电子空穴复合速度越慢,越有利于光催化反应的进行[31]。在有助催化剂的光催化体系中,由于助催化剂捕获了光生电荷,此时更小的荧光寿命意味着更大的非辐射迁移率,有利于光催化反应的进行[31]。磷掺杂的CdS/P平均荧光寿命比CdS更大,说明由P的掺杂而产生的缺陷捕获了电子,导致CdS/P中的电子与空穴复合的过程受阻,提高了荧光寿命。CdS/P/MoS2的平均寿命为1.88 ns,大幅低于CdS/P(126.68 ns)与CdS(4.66 ns)的平均寿命,证明CdS/P/MoS2具有更高的非辐射衰减迁移率,这归功于电子从CdS/P向MoS2的转移。
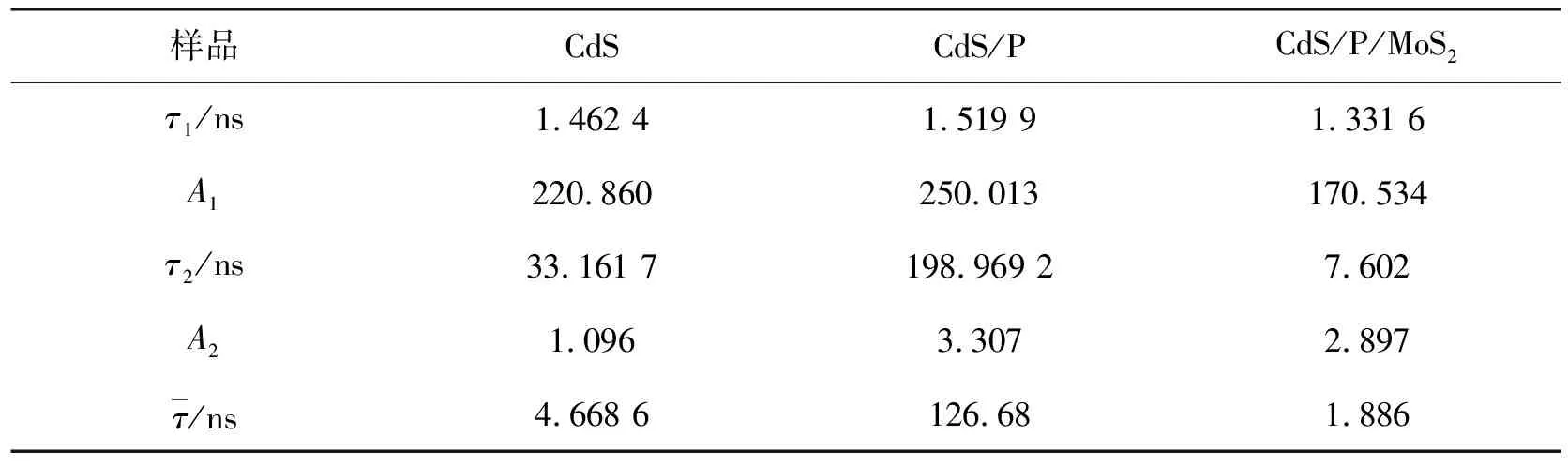
表1 CdS、CdS/P和CdS/P/MoS2的荧光寿命拟合参数
交流阻抗图谱可用于分析样品的界面电阻。在奈奎斯特图中,较小的弧线代表较小的界面电阻,意味着电荷可以更容易从样品表面转移到溶液中[39]。助催化剂的存在有利于将催化剂中的光生电荷传导到溶液中。从图13中可以发现,CdS/P/MoS2的弧线比CdS/P的更小,证明MoS2的存在有利于电荷传导,有利于光催化反应的进行。
瞬态光电流响应可以测试样品抑制光生电子与光生空穴复合的能力。如图14所示,当样品受到间歇的光照时,CdS/P的电流密度显著大于CdS的电流密度,CdS/P/MoS2的光电流比CdS/P显著增强。这说明CdS/P与MoS2之间的界面相互作用导致光生电子可以从CdS/P向MoS2快速迁移,并且由于CdS/P/MoS2具有更小的界面电阻,因此CdS/P/MoS2具有更好的性能。上述光学与光电化学测试同时证明以MoS2作为助催化剂与进行磷掺杂的CdS/P/MoS2具有更好的光吸收能力,同时抑制了光生电荷的复合,并促进电子与空穴向催化剂表面的迁移,从而提高了光催化能力。
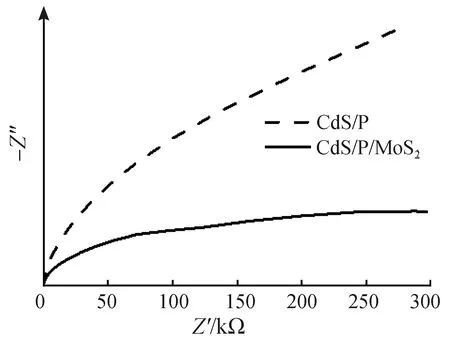
图13 CdS/P与CdS/P/MoS2的电化学阻抗Fig.13 EIS of CdS/P and CdS/P/MoS2
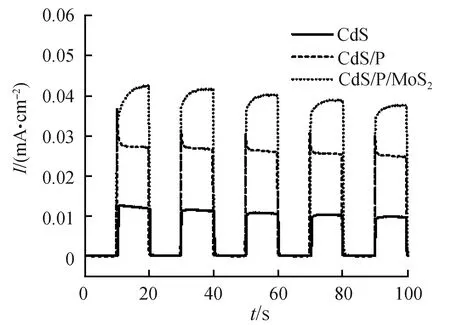
图14 样品的瞬态光响应图Fig.14 Transient photocurrent response curves
为了进一步证明助催化剂MoS2对样品的光催化产氢能力的影响,测试了CdS/P与CdS/P/MoS2的电催化产氢活性和电催化产氧活性。这种测试可用于表征其光催化产氢反应活性和光催化产氧活性[40]。图15为使用线性扫描伏安法测试所得的CdS/P与CdS/P/MoS2的电催化HER曲线和电催化OER曲线,当CdS/P的电流密度达到-0.5 mA/cm2时,电势为-0.751 V,当CdS/P/MoS2的电流密度达到-0.2 mA/cm2时,电势为-0.653 V。由此可见,通过MoS2的负载可以显著降低HER的超电势。从图16中可以看出,当CdS/P的电流密度达到1 mA/cm2时,电势为2.383 V,当CdS/P/MoS2的电流密度达到1 mA/cm2时,电势为2.07 V。由此可见,MoS2的负载可同时降低OER的超电势。
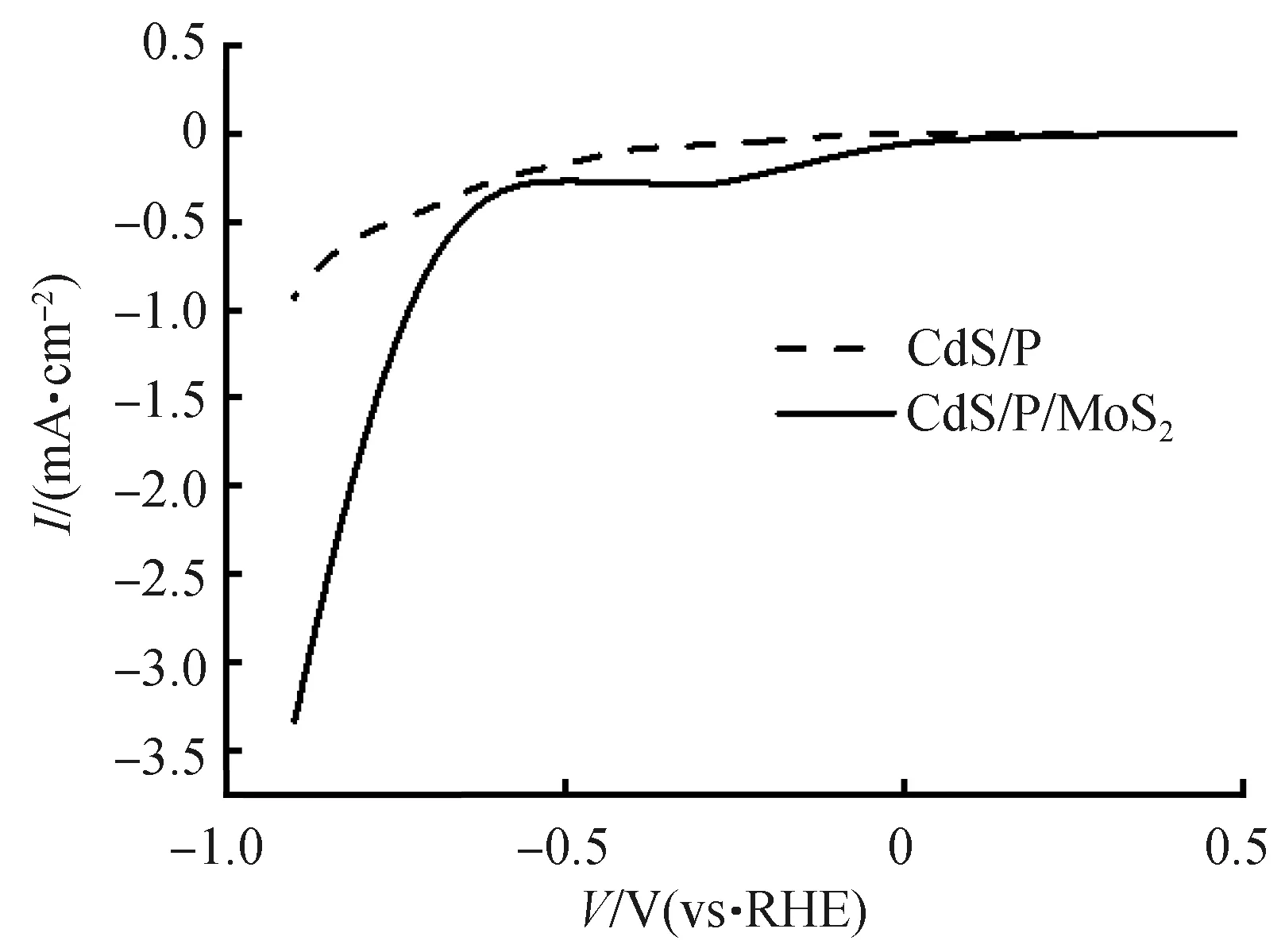
图15 CdS/P与CdS/P/MoS2的HERFig.15 HER curves of CdS/P and CdS/P/MoS2
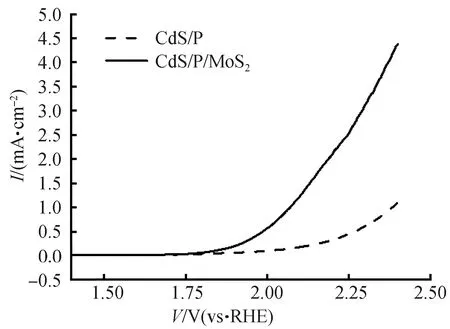
图16 CdS/P与CdS/P/MoS2的OERFig.16 OER curves of CdS/P and CdS/P/MoS2
综合以上实验现象,一个合理的CdS/P/MoS2光催化分解纯水产氢的步骤被推测出来(图17)。在光的激发下,CdS/P中产生了光生电子与空穴。在肖基特能垒的作用下,光生电子从CdS/P中传导到MoS2纳米片表面,而光生空穴被肖基特能垒传导到CdS/P的表面,由此光生电子与空穴相互分离。由于MoS2具有降低HER超电势和OER超电势的作用,MoS2中的光生电子和CdS/P中的光生空穴更易于传导到水中,H2O中的H+与光生电子结合生成H2。综上所述,CdS/P/MoS2光催化剂实现了在纯水中光催化产氢。
最后将CdS/P/MoS2的光催化水解产氢性能与近些年其他CdS相关体系进行对比,如表2所示。CdS/P/MoS2体系并不具有最高的产氢速率,且其在420 nm处的AQE并没有特别理想,但仍然具有一定的应用潜力。
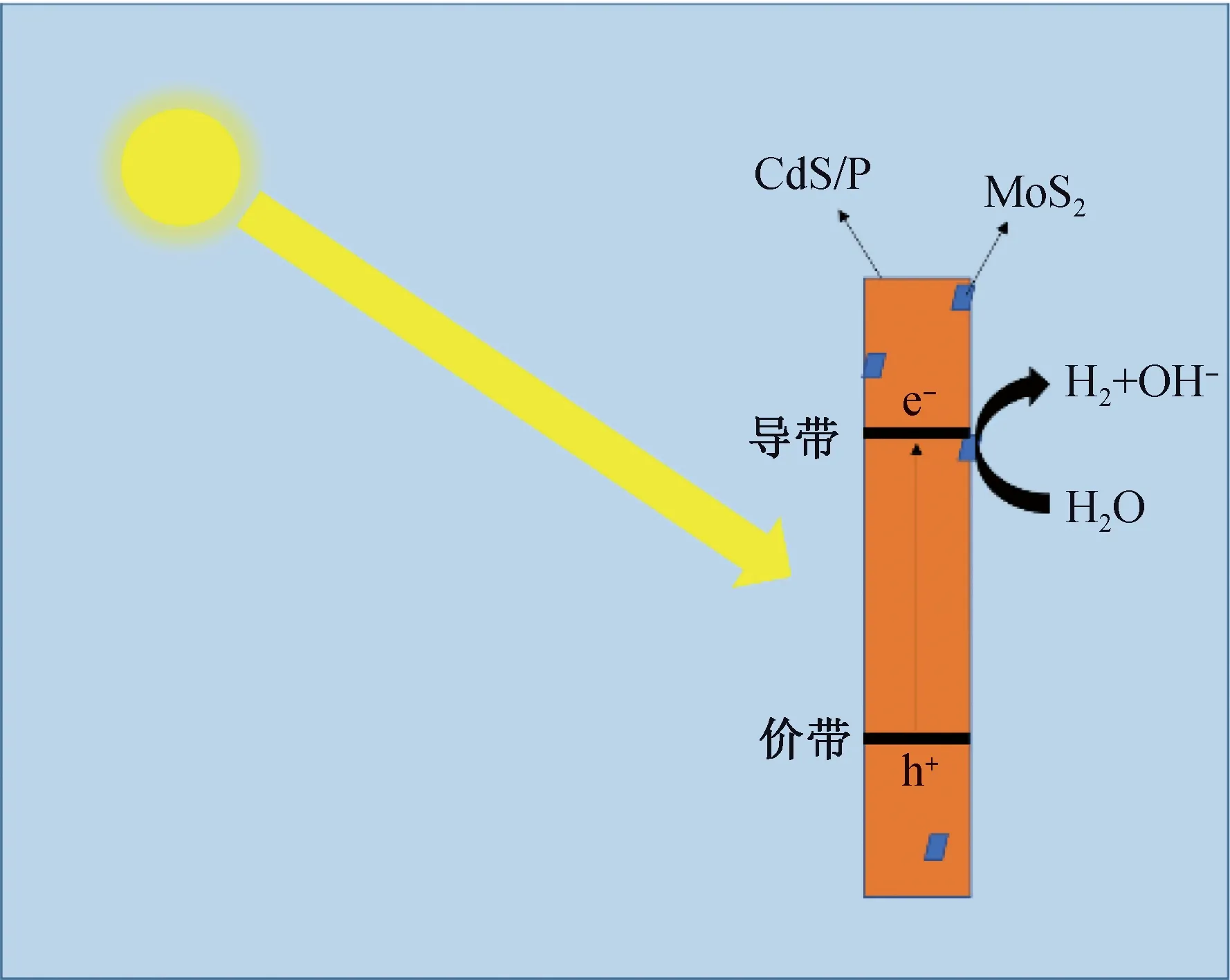
图17 CdS/P/MoS2的光催化水解产氢机理Fig.17 Mechanism of water splitting over CdS/P/MoS2 under visible illumination

表2 近年的CdS相关体系水解产氢性能对比
3 结语
综上所述,P的存在有助于扩大CdS的光响应范围,且有利于阻碍光生电子与空穴的复合。作为一种产氢助催化剂,MoS2有利于降低HER超电势和OER超电势,从而有利于将光生电荷传导到水中生成H2。在P和MoS2的共同作用下,光催化剂CdS/P/MoS2具有了在纯水中光催化产氢的能力。值得注意的是,在光催化实验过程中没有检测到明显的O2产生,这是由于体系中缺少一种高效的析氧助催化剂,推测在光催化过程中产生了过氧化氢或其他氧化物储存在水中,难以检测。CdS/P/MoS2光催化剂具备在纯水中光催化产氢的能力,因此其在将太阳能转化为氢能的应用上具有巨大潜力。为了改进CdS/P/MoS2光催化体系,更进一步提高其光催化能力,有必要对其光催化氧化半反应进行更细致地研究,在CdS/P/MoS2体系中添加合适的析氧助催化剂将是进一步提高其性能的关键手段。
猜你喜欢
物理化学学报(2024年11期)2024-11-06 00:00:00
应用数学和力学(2024年1期)2024-01-29 08:32:46
车用发动机(2021年5期)2021-10-31 05:48:38
电源技术(2021年7期)2021-07-29 08:35:24
——潘桂棠光生的地质情怀
沉积与特提斯地质(2021年2期)2021-07-20 06:33:26
无机盐工业(2017年5期)2017-05-25 00:37:34
物理化学学报(2017年3期)2017-03-11 00:25:30
化工管理(2017年25期)2017-03-05 23:32:36
昭通学院学报(2016年5期)2016-02-24 10:51:12
石家庄铁道大学学报(自然科学版)(2015年3期)2015-02-28 15:05:43
