
钴掺杂锗团簇CoGen-/0(n=2~11)的结构与稳定性研究
2022-10-16 05:21杨欣竹董彩霞杨桔材
当代化工研究 2022年18期
*杨欣竹 董彩霞 杨桔材*
(1.内蒙古工业大学 能源与动力工程学院 内蒙古 010051 2.内蒙古工业大学矿业学院 内蒙古 010051)
团簇作为一些材料的构筑基元,在实验和理论方面已经开展了大量研究,使得它们在磁性纳米材料、微电子行业和环境催化等领域具有潜在应用。近些年硅团簇及硅掺杂金属团簇的研究日趋成熟[1-5],寻求高稳定性,高传输速度,耐热的团簇构筑基元,成为半导体团簇领域的研究热点,由于锗的电子和空穴迁移率比硅高,研究人员逐渐将视野转移到锗元素[6]。对半导体团簇领域而言,由于锗原子容易通过sp3杂化相互作用,使得团簇表面存在悬挂键,所以纯锗不能形成稳定的笼型结构。研究发现,锗团簇中掺杂过渡金属原子可以吸收锗团簇中的悬挂键,可以得到过渡金属原子位于中心的稳定的笼型结构,进而产生新颖的物理化学特性。这使得内嵌过渡金属原子的笼型锗团簇可以成为很好的团簇自组装材料基元[7-10]。关于过渡金属掺杂锗团簇的研究已有很多,其中钴掺杂锗团簇的研究也引起了科学家们的兴趣。例如:郑卫军团队[11]使用光电子光谱实验方法研究了CoGen-(n=2~11),理论上采用B3PW91密度泛函和6-311+G(d)基组系统地研究了中性和阴离子团簇的电荷和磁性等性质。Jing等人[12]采用mPW1PW91泛函结合6-311+G基组,研究了CoGen(n=2~13)的低能量构型及性质。Zhang等人[13]利用激光气化装置和质谱研究了CoGe10阴离子团簇的构型。Uţă等人[14]利用B3LYP密度泛函结合6-31G(d)基组计算了CoGe10z(z=-5~+1)基态结构及对应的电子态。Trivedi等人[15]利用B3LYP方法结合LANL2DZ基组计算了M@Ge12(M=Co,Pd,Tc和Zr)的结构和稳定性,电荷转移结果表明钴原子与其他金属不同,更倾向于作为电子受体。Kapil和他的团队[16]运用PW91密度泛函方法结合平面波基组研究了过渡金属掺杂锗团簇TMGen(TM=Mn,Co,Ni;n=1~13),报道了团簇的基态结构,系统地研究了过渡金属掺杂产生的性质。Tran等人[17-18]运用CASPT2和RASPT2方法研究了CoGen-/0(n=1~3)和CoGen-(n=4~5)的结构及相关性质。综上所述,关于Co掺杂锗团簇的理论研究已有很多,除了Tran等人使用了CASPT2和RASPT2方法对小尺寸团簇(n≤5)进行了研究外,其他研究所采用的计算方法均为单杂合密度泛函方法。而对于含过渡金属原子体系,由于自旋等因素导致多个势能面交叉,即使对真正的基态也存在着强烈的多参考组态相关的问题,这是单杂合密度泛函方法解决不了的问题。多参考组态方法(如MCSCF、CASPT2)只适合小分子体系的计算。另外,已有文献报道在选取初始构型时并未采用全局搜索方法,只有人工设计的初始构型,构型不够全面,可能会错失真正的基态构型,导致预测的基态构型不准确。因此,本文基于无偏见的全局搜索方法结合高精度的耦合簇方法CCSD(T)对CoGen-/0(n=2~11)团簇的基态结构与稳定性进行了系统的研究。
1.计算方法
初始构型的选取使用ABCluster全局搜索技术[19],选用PBE0单杂合密度泛函方法结合LANL2DZ有效核赝势基组,对每个尺寸的CoGen-/0(n=2~11)团簇随机产生400个结构,选择能量差在0.8eV内的结构,进行初始构型的选取;接着使用PBE0方法,对钴原子和锗原子均使用cc-pVTZ全电子基组进行构型初步优化,并在同一水平上计算了振动频率,以确保优化的几何结构是势能面上真正的全局最小点,所有的结构优化都不加任何的对称性约束。再从初步优化结果中选取低能量的构型,提高优化精度,使用双杂合密度泛函(mPW2PLYP)方法,依然采用cc-pVTZ全电子基组,对团簇进行进一步优化,由于在PBE0方法得优化中已计算了震动频率,所以在这一步优化时,为了节约时间,mPW2PLYP方法下的团簇结构优化不做频率计算。对于阴离子团簇的电子态我们考虑了1态、3态和5态;对于中性团簇的电子态考虑了2态、4态和6态。为确保基态结构的准确性,选择双杂合密度泛函(mPW2PLYP)优化后的构型,使用CCSD(T)高精度方法[20]结合cc-pVTZ-DK[21-22]基组计算了结构的单点能,在缺乏实验的情况下,CCSD(T)方法计算的结果可以代替实验值。本文所有计算都在Gaussian 09软件包[23]中完成。
2.结果与讨论
(1)CoGen-/0(n=2~11)团簇的基态结构
通过上述方法计算了钴掺杂锗团簇阴离子和中性的基态构型,分别如图1和图2所示,同时列出了团簇的对称性和电子态。
对于阴离子团簇,我们计算得到CoGe2-团簇的基态构型为等腰三角形,具有C2v对称性和3B1电子态,它可以看作一个钴原子取代Ge3-团簇[6]上的一个锗原子得到的构型。CoGe3-的基态结构是平面菱形结构,对称性为C2v,电子态为3B1,可以看作一个钴原子取代Ge4-团簇[6]上的一个锗原子得到的构型。CoGe4-具有3A''电子态,对称性为Cs,构型为扭曲的四棱锥,可看作由钴原子取代Ge5-团簇[6]中的一个锗原子而来。CoGe2-、CoGe3-和CoGe4-团簇的基态结构及电子态与Tran[17-18]报导的结果一致。5A-1的对称性为C2v,电子态为3B1,可以看作一个钴原子取代Ge6-团簇[6]上的一个锗原子,为四角双锥构型。与郑卫军等人[11]和Tran报导[18]的基态构型及电子态不同。6A-1为钴原子在顶点的五角双锥构型,可以看作一个钴原子取代Ge7-团簇上的一个锗原子[6],对称性为C5v,电子态为1A1。7A-1的对称性为Cs,电子态为3A'',其构型可以描述为钴原子取代Ge8-团簇[6]上一个锗原子而得,可看作五角双锥盖帽一个锗原子结构。8A-1具有Cs对称性,电子态为3A'',可以看作一个钴原子取代Ge9-团簇[6]上一个锗原子得到的构型,可描述为五角双锥盖帽两个锗原子结构。当n=9时形成具有C3v对称性的半笼结构,电子态为3A1。10A-1是由两个五边形和五个四边形将钴原子包裹起来的结构,即TPFQ结构,这时钴原子完全被封装在锗笼里,对称性为C2v,具有1A1电子态,与Zhang报导[13]的基态结构不同。CoGe11-的基态结构为锗笼构型,可以看作由一个锗原子吸附在10A-1结构上而来,具有Cs对称性,电子态为1A'。
对于中性团簇,除n=3,6和11外,基态构型与对应的阴离子团簇的基本相似。CoGe2团簇的基态构型的对称性为C2v,电子态为4B1,它可以看作是一个钴原子取代Ge3团簇[6]上的一个锗原子而来。CoGe3的基态结构为具有C3v对称性和4A'电子态的四面体构型。CoGe4团簇的基态结构具有2A''电子态和Cs对称性,可以看作是钴原子取代Ge5团簇[6]上的一个锗原子而来。CoGe5团簇基态构型的对称性和电子态分别为C2v和4B1,可以看作一个钴原子取代Ge6团簇[6]上一个锗原子而得到的构型。CoGe6的基态构型为钴原子在棱上的五角双锥构型,具有Cs对称性和4A'电子态,其构型可以描述为一个钴原子取代Ge7团簇[6]上一个锗原子而得。基态构型与CoGe7-构型相似的7N-1具有Cs对称性和4A''电子态,其构型可以描述为钴原子取代Ge8团簇[6]上一个锗原子而得到。CoGe8团簇的基态结构具有Cs对称性,电子态为4A'',可以看作三棱柱盖帽两个锗原子和一个钴原子结构。对于CoGe9,基态构型具有C3v对称性和4A1电子态的,它可以看作由8N-1上多吸附了一个锗原子而来,此时钴原子有向锗笼内部包裹的趋势,形成半笼型结构。CoGe10具有C2v对称性和2B1电子态,是由两个五边形和五个四边形将钴原子包裹起来的结构,即TPFQ结构,这此时钴原子完全内嵌在锗笼内部,团簇形成笼型结构。CoGe11构型可看作为1-4-4-2结构,对称性为C2v,电子态为2A1,与郑等人[11]报导的基态构型不同。
(2)CoGen-/0(n=2~11)团簇的热力学稳定性
为了解CoGen-/0(n=2~11)团簇的热力学稳定性,计算了它们的平均键能(ABE)和二阶能量差分(Δ2E)。定义如下:

其中,E代表在CCSD(T)理论水平下计算得到的总能量;E(Ge)代表Ge原子的能量;E(Co)代表Co原子的能量;E(CoGen-/0)代表阴离子和中性团簇的能量。平均键能(ABE)揭示了在团簇之间相对稳定性的关系,平均键能越高,表明团簇越稳定。二阶能量差分(Δ2E)反映了一个团簇与其相邻团簇之间的相对稳定性。二阶能量差分(Δ2E)值越大,说明团簇的相对稳定性越好。CoGen-/0(n=2~11)团簇的平均键能和二阶能量差分变化趋势如图3和图4所示。从图3可知:①钴掺杂锗团簇的阴离子和中性的平均键能的变化趋势相同,平均键能的值整体趋势都随着锗原子数目的增加而增大。②阴离子团簇平均键能均明显比中性团簇高,说明中性团簇在得到一个电子后团簇的稳定性得到了显著的提高,表明在阴离子团簇中,钴原子与锗簇之间的相互作用强于相应的中性团簇,原因可能是额外的电子与锗簇悬挂键相互结合,从而增强了稳定性。③CoGe10-阴离子团簇的平均键能最大。

图3 CoGen-/0(n=2~11)团簇基态结构的平均键能(ABE)
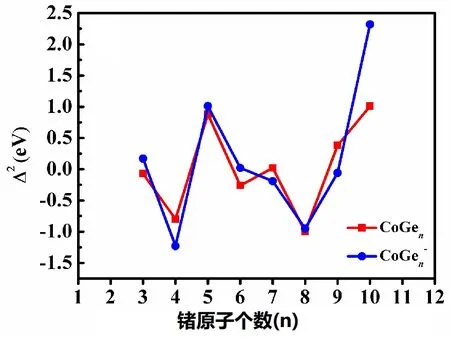
图4 CoGen-/0(n=2~11)团簇基态结构的二阶能量差分(Δ2E)
二阶能量差分的结果表明(见图4):①对于中性团簇,在n=3~9,CoGen中性团簇的二阶能量差分呈奇偶交替变化;②CoGen中性团簇和CoGen-阴离子团簇在n=3、5、10时,稳定性高于相邻团簇,即稳定性较高,而在n=4和n=8时稳定性较低;③在n=10时,阴离子团簇的二阶能量差分值明显高于中性和其他尺寸的值。综上所述,闭壳层电子结构的CoGe10-阴离子团簇具有良好的热力学稳定性,有望成为新型多功能环保材料的合适的结构单元。
3.结论
采用高精度的CCSD(T)方法对过渡金属钴掺杂锗团簇阴离子及中性团簇CoGen-/0(n=2~11)的结构进行了优化,确定了基态结构,同时,计算和分析了阴离子及中性团簇的平均键能和二阶能量差分来预测团簇的热力学稳定性,进而了解掺杂团簇的性质。在构型演化方面,结果表明,阴离子团簇在n≤8时,基态结构的生长模式是取代结构;当n=9时,团簇形成半笼结构;n=10是团簇的最小成笼尺寸,即钴原子内嵌入锗笼内部。中性团簇在n≤7时,基态结构的生长模式是取代结构;同阴离子团簇相同,当n=9时,团簇形成半笼结构;n=10以后形成笼型结构。除了CoGe3、CoGe6和CoGe11团簇外,中性团簇和阴离子团簇的基态结构基本相似。在性质分析方面,平均键能与二阶能量差分分析表明,团簇在n=10时,阴离子团簇的稳定性明显高于其他尺寸的团簇。所以CoGe10-具有良好的热力学稳定性,有望作为组装材料的基本单元,用来制备光电材料、光敏材料或催化等新型多功能环保材料。
猜你喜欢
初中生学习指导·中考版(2022年4期)2022-05-12
航空动力(2022年2期)2022-05-06
中国药房(2022年7期)2022-04-14
安徽工业大学学报(自然科学版)(2021年3期)2021-09-08
中学课程辅导·高考版(2020年9期)2020-10-20
智富时代(2019年4期)2019-06-01
智富时代(2019年4期)2019-06-01
科学与财富(2016年28期)2016-10-14
中学生数理化·八年级数学人教版(2016年6期)2016-08-22
读写算·小学中年级版(2016年5期)2016-05-14
