
华北亚高山土壤细菌群落及氮循环对退耕还草的响应
2022-10-13 05:14王礼霄刘晋仙柴宝峰
生态环境学报 2022年8期
王礼霄,刘晋仙,柴宝峰
山西大学黄土高原研究所/黄土高原生态恢复山西省重点实验室,山西 太原 030006
亚高山草地具有水源涵养、生物多样性保育、水土保持和碳汇功能(赵鹏宇,2019)。受全球变化和人类活动的影响,山西亚高山草地植被退化,水土流失,生态系统功能严重受损。基于生态环境的保护和改善,我国实施了“退耕还林/草”工程(常庆瑞等,1999)。山西亚高山草甸面积约33.33×104hm2,占全省草地总面积的10%。这一重要的政策实施以来,植被得以迅速恢复。植物是生态系统结构和功能自然演化的主要驱动力(史利江等,2021),凋落物和根系分泌物的输入影响土壤的理化性质(Kuypers et al.,2018),进而驱动了土壤微生物群落的演替过程(Kardol et al.,2010;Bardgett et al.,2014)。亚高山生态系统对气候变化敏感,恢复较为困难,植被和土壤微生物群落的演替规律尚不清楚。
目前关于退耕还草地恢复过程中植物多样性和土壤特征的变化规律已有了一定的研究。随着演替的进行,植物生物量和物种多样性会随着时间的推移而增加(樊博等,2020)。自然植被的恢复可以改善退化土壤的性质并增加肥力(Cao et al.,2021)。植物演替对土壤养分有积极的作用,并增强了植物的多样性(Deng et al.,2014;Matetskaya et al.,2021)。Wang et al.(2011)发现黄土高原农田弃耕后,土壤有机碳、全氮、速效氮和速效钾以及土壤酶活性随着植被恢复年限的增加而提高。
土壤微生物在生态系统物质循环过程中发挥着关键作用,微生物群落结构和功能的变化会影响植物残体分解、土壤碳氮转变和地上植被养分供给等生态过程(Wang et al.,2011)。同时,微生物多样性、结构和功能也受到植被、土壤特性等因素的影响(Fan et al.,2016;Bier et al.,2014)。有越来越多的研究关注于土壤微生物群落对地上植被演替的响应,地上植被的演替过程会对土壤微生物类群和功能多样性等造成重要影响(Lee-Cruz et al.,2013;Nacke et al.,2014)。Li et al.(2014)和Zuo et al.(2016)对植被恢复矿区和沙地草原土壤微生物群落的分析发现,细菌的多样性随着植被恢复时间的增长而显著提高,呈现出明显的时间演替趋势。多样性的增加对微生物群落的功能产生重要的影响,功能基因丰度可以反映微生物群落的功能对环境变化的响应(Li et al.,2014)。目前关于地上植被如何影响土壤微生物氮循环功能仍有争议,Blaud et al.(2017)报道,在从农田到森林的次生演替过程中,氮循环相关基因的丰度增加;而Zhong et al.(2018)观察到,在从草原到森林的次生演替过程中,氮循环相关基因的相对丰度则是先升高后降低。因此,退耕还草过程中植物群落与微生物群落结构、多样性和功能之间的关系是恢复生态学有待回答的问题。
本研究以山西省宁武云中山亚高山退耕还草区不同恢复年限(15、20、30 a)的草地为研究对象,进行草地植物群落调查和土壤样本的采集,利用16S rRNA基因的Illumina MiSeq测序和qPCR技术,通过多元统计的方法分析草甸恢复过程中土壤细菌群落的结构与功能特征及其驱动因素,阐明土壤细菌群落对地上植被恢复时间序列的响应。本研究旨在回答以下问题:(1)亚高山草草甸退耕还草过程中,植物多样性、土壤性质和土壤细菌群落的演变规律;(2)退耕还草过程中土壤细菌群落结构和氮循环功能演变的驱动机制。
1 材料与方法
1.1 样品采集
研究地点位于山西省吕梁山脉北段分支宁武云中山,呈东北—西南走向,海拔约 2160 m。本区年平均气温2.0—9.0 ℃,无霜冻期84—135 d,年降水量430—700 mm。该地区自上世纪90年代开始实施退耕还草措施,农田植被开始自然恢复。1个农田样地(对照)和3个不同恢复年限的草甸(15、20、30 a)作为试验样地。样地具有相似的坡度和海拔,并经历了类似的耕作方式。退耕前,农田的主要作物是莜麦,退耕后,植被自然演替。选择对照农田种植的也是莜麦,在春季施用了农家肥。
在每个样地,设置3个1 m×1 m的样方,样方之间间隔约20 m。用土钻采集表层土壤样品(0—10 cm),每个样方内采集5个子样(每个角1个,中心1个),在聚乙烯袋中混合成一个样品。经2 mm网筛,将大部分根、动物和石头去除,然后将样本分成两部分。其中一部分风干后进行土壤理化性质测定,另一部分保存在-80 ℃冰箱中,用于DNA的提取。在每个样方中调查植被参数,记录每种植物的名称、高度、盖度及多度等指标。
1.2 土壤理化性质测定
烘干法测定土壤含水量(SWC);土壤pH值用电位法(HANNA,意大利)测定(土水质量比为1∶2.5);总碳(TC)和总氮(TN)通过元素分析仪(ElementarVario MACRO,德国)测定;铵态氮(NH4+-N)、硝态氮(NO3--N)采用间断元素分析仪(CleverChem 380,德国)测定。
脲酶采用苯酚钠-次氯酸钠比色法,其活性以37 ℃下培养24 h后1 g土壤产生的NH3的mg数表示;蔗糖酶采用3,5-二硝基水杨酸比色法,其活性以37 ℃培养24 h后1 g土壤产生的葡萄糖的mg数表示;过氧化氢酶活性(H2O2)采用KMnO4滴定法,其活性以20 min内每克土壤分解的过氧化氢的毫克数来表示。
1.3 DNA提取、高通量测序及生物信息学分析
称取0.5 g土壤样品,使用E.Z.N.A.土壤DNA试剂盒(Omega Bio-tek,USA)提取和纯化土壤微生物DNA。将每个采样点的3个土壤样品等体积混合,一共12个DNA样品,送往上海美吉生物医药科技有限公司进行高通量测序。采用338F(正向引物 5′-ACTCCTACGGGAGGCAGCA-3′)和806R(反向引物 5′-GGACTACHVGGGTWTCTAAT-3′)对细菌16S rRNA的V3—V4高可变区进行PCR扩增,并通过Illumina Miseq测序平台对扩增产物进行测序。剔除嵌合序列后,剩下的序列使用UPARSE以97%的相似性作为阈值划分分类操作单元(operational taxonomic units,OTUs)。
1.4 氮循环功能基因的定量PCR
采用CFX 96 touch实时定量 PCR系统(Bio-Rad,Hercules,CA,USA)测定参与土壤氮循环过程的主要功能基因(nifH、amoA-AOA、amoA-AOB、nirK、nirS)丰度。操作过程和引物序列见课题组之前的报道(Luo et al.,2020)。反应体系、扩增程序及熔解曲线见文献(徐白璐等,2017)。以含有目的基因片段的重组质粒为模板,10倍倍比进行稀释,根据情况选取4—5个稀释梯度制作标准曲线。将样品与标准品同时进行 qPCR检测,每个样品设置 3个重复和一个阴性对照。qPCR的扩增效率是 80%—95%,标准曲线的R2>0.990。所有功能基因的丰度最终计算为每克干土的拷贝数。
1.5 数据分析
基于R studio(v.3.4.3)vegan包,计算细菌和植物的α多样性和β多样性,采用非度量多维尺度分析(Non-metric multidimensional scaling,NMDS)和相似性分析(Analysis of similarity,ANOSIM)对农田与不同恢复年限草地细菌群落结构进行比较。采用冗余分析(Redundancy analysis,RDA)评价细菌群落结构与环境变量之间的相关性。利用IBM SPSS statistics 20进行皮尔逊相关性分析(Pearson correlation test)以及采用单因素方差分析(One-way analysis of variance)和Duncan多重比较分析进行显著性差异分析。所有统计分析的显著性水平均为P<0.05。
2 结果
2.1 土壤理化性质和植物多样性特征
不同恢复年限草地的土壤理化参数和植物多样性数据见表1。所有土壤样品的pH都小于7,为弱酸性土壤。土壤总碳、总氮、碳氮比、硝态氮、铵态氮、土壤含水率、过氧化氢酶、脲酶和蔗糖酶活性在农田和不同年限恢复年限的草地间差异显著(P<0.05),而土壤pH的差异不显著(P>0.05)。农田土壤总碳、总氮、铵态氮、硝态氮含量、脲酶活性显著高于草地,而在草地恢复过程中土壤总碳和总氮含量稳步提升;退耕30年草地土壤碳氮比、含水率和蔗糖酶活性显著高于其他样地。不同恢复年限的草地地上植被多样性指数(Shannon index)和丰富度指数(Richness index)差异显著(P<0.05),但均匀度差异不显著(P>0.05)。恢复15年的草地植物生物多样性最高,随着恢复年限的加长,草地优势种发生变化,生物多样性降低。
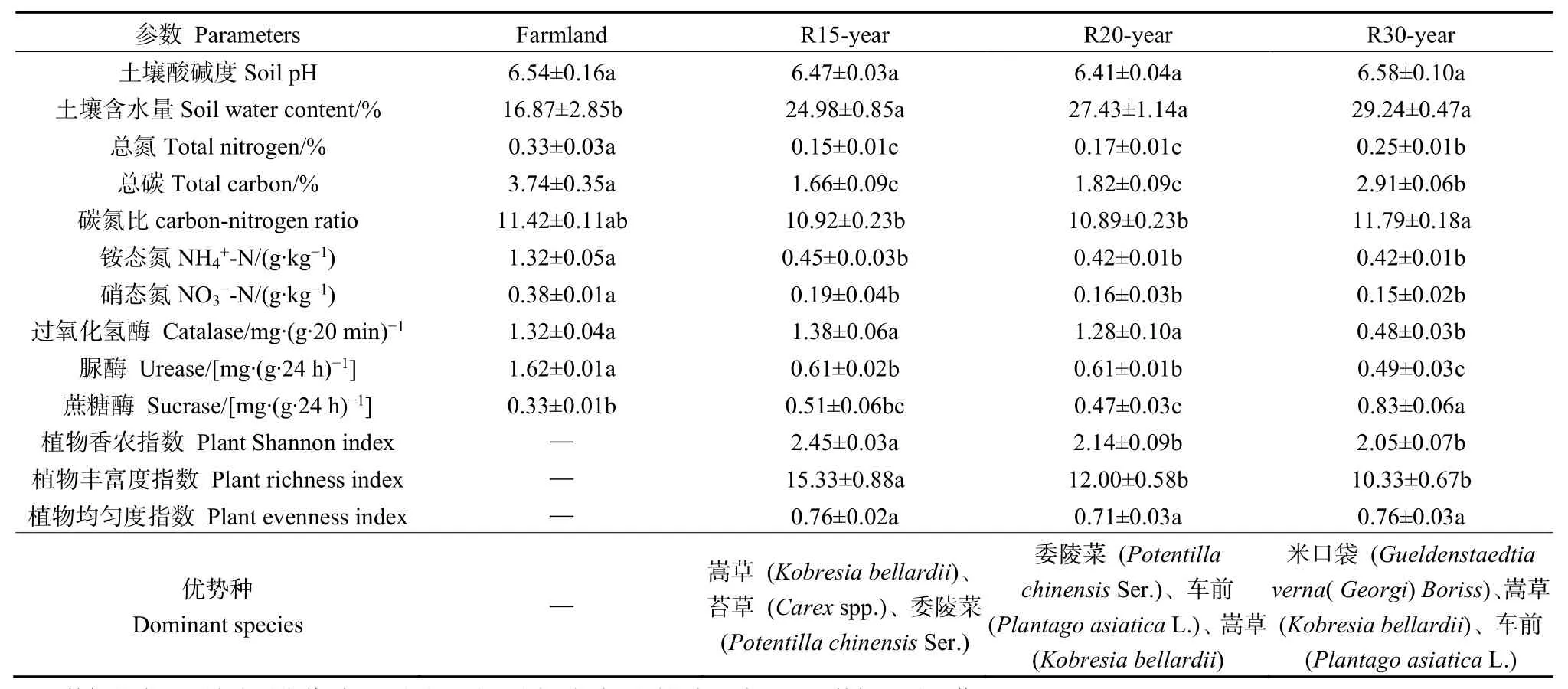
表1 农田与不同恢复年限草地土壤理化性质与植物群落参数Table 1 Soil physicochemical properties and plant community parameters under different revegetation habitats and farmland
2.2 细菌群落组成和多样性
通过高通量测序,在12个样本中共检测到358488个细菌序列。按最小样本序列数抽平,共鉴定出5410个OTU(>97%序列相似性水平),属于36个门、118个纲、295个目、449个科、808个属和1709个种。36个门中有11个门定义为优势细菌门(样本中相对丰度>1%),相对丰度从高到低,依次为放线菌门(Actinobacteria)、变形菌门(Proteobacteria)、酸杆菌门(Acidobacteria)、绿弯菌门(Chloroflexi)、芽单胞菌门(Gemmatimonadetes)、厚壁菌门(Firmicutes)、疣微菌门(Verrucomicrobia)、拟杆菌门(Bacteroidetes)、髌骨细菌门(Patescibacteria)、甲基肌酐门(Methylomirabilota)和粘球菌门(Myxococcota)(图1a)。在不同恢复年限退耕草地的土壤中,这些优势细菌门占据了95%以上的细菌序列。绿弯菌门的相对丰度在不同恢复年限退耕草地之间存在显著差异(P<0.05)(图1b)。
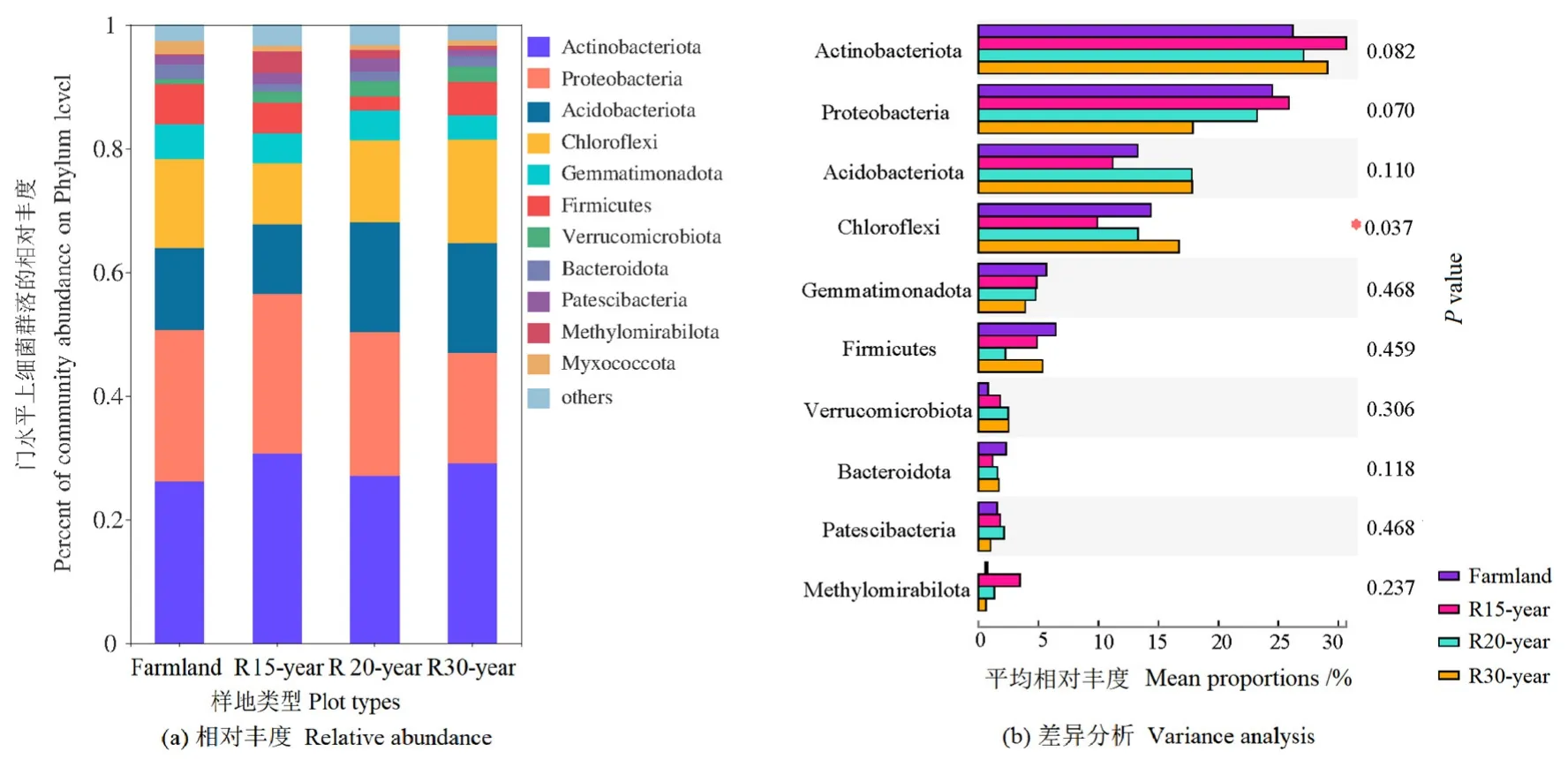
图1 农田和不同退耕年限草地细菌群落在门水平的相对丰度及差异分析Figure 1 The relative abundance of dominant bacteria phylum and variation analysis from different revegetation habitats and farmland
12个相对丰度最高的细菌纲、科和属,在亚高山不同恢复年限退耕草地间存在差异(图2)。α变形菌纲(Alphaproteobacteria)、γ变形菌纲(Gammaproteobacteria)、Blastocatellia、绿弯菌纲(Chloroflexia)的丰度差异显著;在科水平,黄色杆菌科(Xanthobacteraceae)、Pyrinomonadaceae、Roseiflexaceae的差异显著;RB41和norank_f_Roseiflexaceae属在不同恢复年限退耕草地间存在显著差异(P<0.05)。
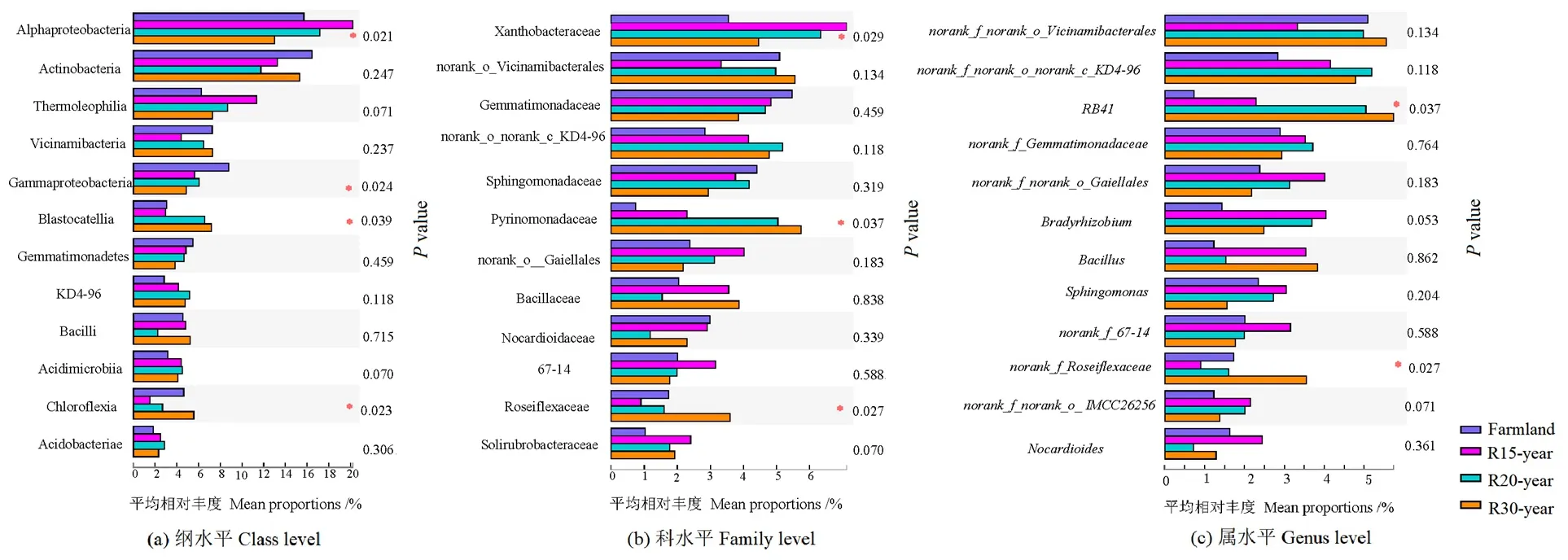
图2 农田和不同退耕年限草地细菌群落在纲、科、属水平的差异分析Figure 2 The variation analysis of dominant bacteria class,family and genus from different revegetation habitats and farmland
不同恢复年限退耕草地的土壤细菌群落α多样性分析结果见表2。其中农田土壤细菌群落Shannon指数显著高于退耕还草地,Simpson指数显著低于退耕还草地,即农田土壤细菌群落多样性最高,而均匀度较低。自然恢复 15年的退耕地的细菌群落Shannon指数显著低于其他样地,Simpson指数显著高于其他样地。随着自然恢复年限的增长,细菌群落Shannon指数增加,Simpson指数降低。农田土壤细菌群落Sobs指数、ACE指数和Chao1指数最大,其土壤细菌群落种类数目最多,在退耕地自然恢复过程中,细菌群落种类数目呈增加趋势。
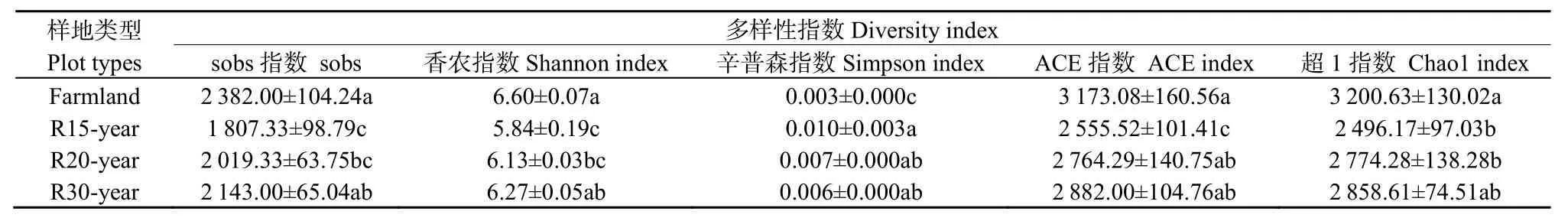
表2 农田和不同恢复年限草地土壤细菌群落多样性Table 2 Diversity of bacteria communities of different revegetation habitats and farmland
基于Bray-Curtis距离的非度量多维尺度分析(NMDS)和ANOSIM结果表明,不同恢复年限退耕草地的土壤细菌群落结构发生了显著的变化(R2=0.5014,P<0.01)(图3a)。统计分析结果显示,细菌群落的Bray-Curtis距离符合正态分布(P>0.05),土壤细菌群落 β多样性(Bray-Curtis距离)与组间时间距离呈显著正相关(r=0.449,P<0.001),土壤细菌群落之间的相异性随着恢复时间的增加而增加(图3b)。
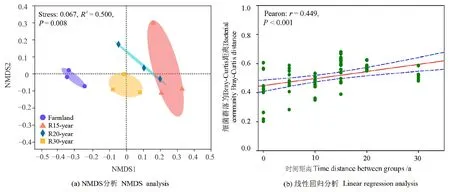
图3 农田和不同恢复年限草地细菌群落基于Bray-Curtis距离的NMDS分析及其与线性回归分析Figure 3 NMDS and linear regression analysis of bacteria communities based on Bray-Curtis distance among different revegetation habitats and farmland
2.3 环境因子对细菌群落的影响
环境因子对不同恢复年限草地细菌群落的组成具有一定的影响。冗余分析(RDA)的前两个轴解释了总变量的48.42%。从图4a可知,环境变量总体上沿着时间序列分布。在环境变量中,总碳、总氮、碳氮比与铵态氮含量是细菌群落结构变化的重要驱动因子(P<0.05)。此外,通过环境因子对细菌主要类群影响的Pearson相关分析发现(图4b),土壤总碳、总氮与绿弯菌门、粘球菌门显著正相关(P<0.01),土壤含水量与变形菌门和蛭弧菌门(Bdellovibrionota)显著负相关(P<0.01),与浮霉菌门显著正相关(P<0.01),土壤铵态氮含量与黏细菌门显著正相关(P<0.01),土壤硝态氮含量与变形菌门、厚壁菌门、拟杆菌门与粘球菌门显著正相关(P<0.01),与浮霉菌门与甲基肌酐门显著负相关。植物群落多样性与土壤细菌群落之间的相关性分析结果表明(图 5),土壤细菌和植物的Shannon指数显著相关(P<0.05),而它们之间的β多样性没有显著的相关关系(r=0.127,P>0.05)。
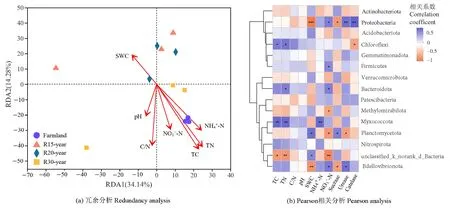
图4 农田和不同恢复年限草地细菌群落组成的冗余分析及优势门与环境因子的Pearson相关分析Figure 4 Redundancy analysis of bacterial community composition and Pearson analysis with dominant phylum in different revegetation grasslands and farmland

图5 农田和不同恢复年限草地土壤细菌群落与植物群落α多样性和β多样性之间的相关关系(基于Bray-Curtis距离)Figure 5 Relationship between plant and soil bacterial α-diversity and β-diversity (based on Bray-Curtis distances)in different revegetation grasslands and farmland
2.4 退耕还草过程中氮循环功能基因的变化特征
利用实时定量 PCR方法分析了土壤中与氮循环有关的关键功能基因,包括固氮有关的nifH基因、与硝化作用有关的amoA-AOA基因、amoA-AOB基因、反硝化作用有关的nirK和nirS基因。结果显示,在4个样地中土壤中nifH的拷贝数是 1.31×108—4.04×108g-1,amoA-AOA 的拷贝数是 0.33×108—2.47×108g-1,amoA-AOB 的拷贝数是0.31×107—7.28×107g-1,nirK 的拷贝数是 3.32×107—4.55×107g-1,nirS的拷贝数是0.28×107—4.30×107g-1(图6)。农田中5种氮循环有关的关键功能基因的拷贝数均显著高于退耕还草自然恢复样地,amoA-AOA、amoA-AOB、nifH、nirS的拷贝数在4个样地间有显著差异(P<0.05),且随着恢复年限的增长,amoA-AOB与 nifH的拷贝数显著增加(P<0.05),nirK的拷贝数在4个样地间无显著差异(P>0.05)。
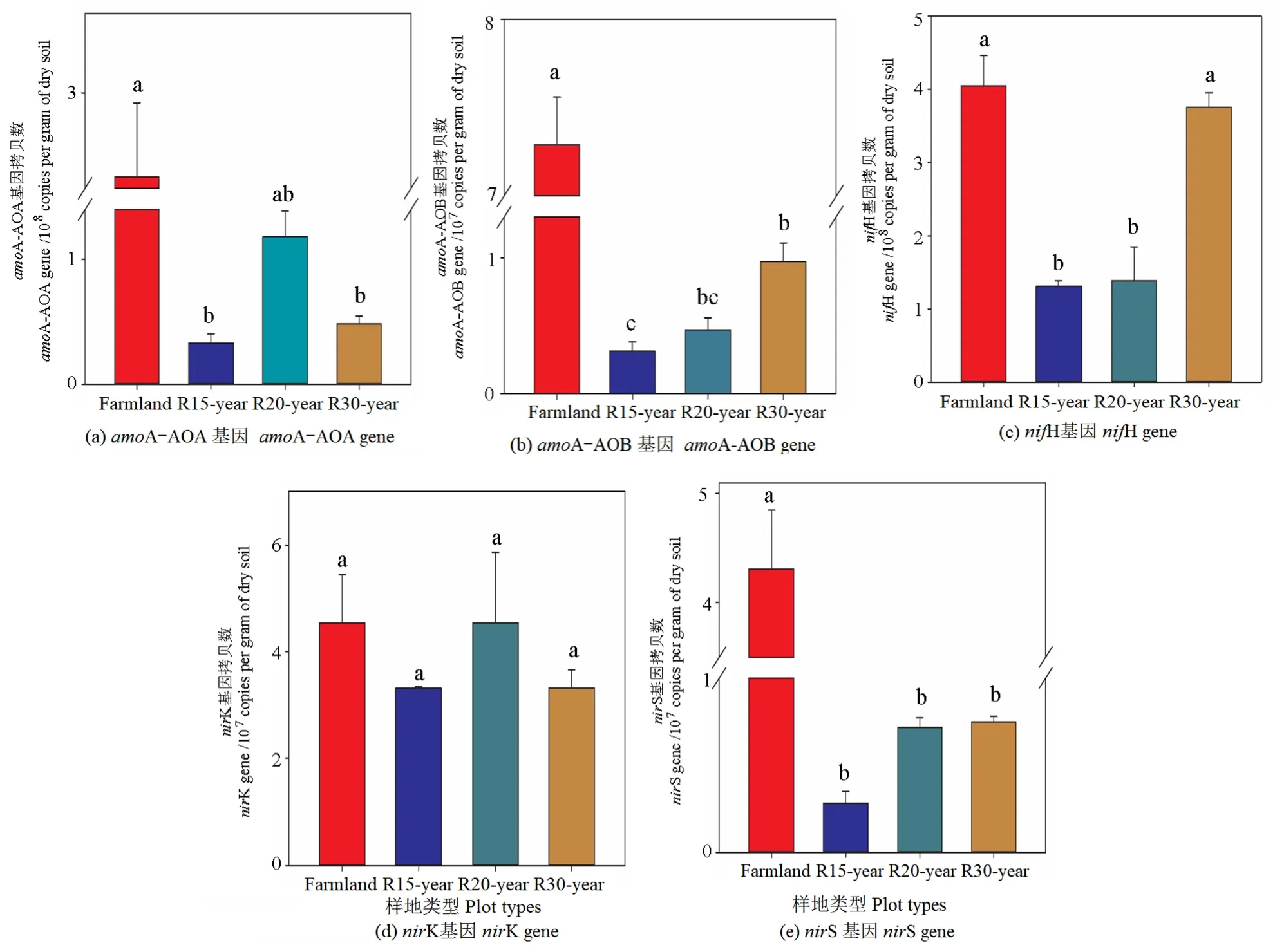
图6 农田和不同恢复年限草地参与氮循环主要过程的功能基因拷贝数(amoA-AOA,amoA-AOB,nifH,nirK and nirS)Figure 6 Gene copy numbers of functional genes involved in major steps of the nitrogen cycle (amoA-AOA,amoA-AOB,nifH,nirK and nirS) in different revegetation grasslands and farmland
2.5 氮循环功能基因与环境参数和优势类群的相关关系
用Pearson相关性分析环境因子对氮循环相关基因拷贝数的影响。编码氨单加氧酶基因amoA-AOA与amoA-AOB对环境因子的响应一致,amoA-AOA与amoA-AOB与总碳、总氮、铵态氮、硝态氮显著正相关,与土壤含水率显著负相关(图7)。编码固氮酶基因 nifH、总碳、总氮、碳氮比与铵态氮显著正相关。编码亚硝酸还原酶基因nirS、nirK对环境因子的响应不同,环境因子与nirK相关性不显著,但nirS与总碳、总氮、铵态氮与硝态氮显著正相关,与土壤含水量显著负相关。
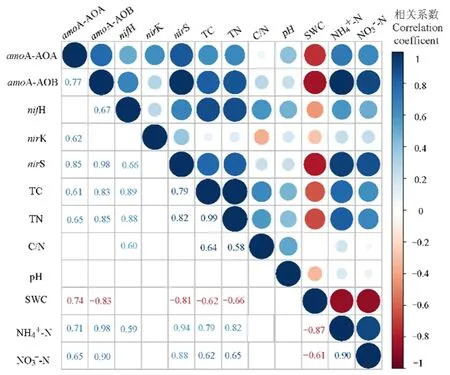
图7 参与氮循环主要过程的功能基因与土壤理化性质的相关关系Figure 7 Correlations between gene copy numbers of functional genes involving in nitrogen cycling and soil physicochemical properties
氮循环相关基因与细菌优势门的相关性(表3)表明,氮循环相关基因与优势门具有一定的相关性。其中,amoA-AOA基因与绿弯菌门和拟杆菌门极显著正相关(P<0.01),与粘球菌门显著正相关(P<0.05);amoB-AOB基因与绿弯菌门、拟杆菌门和粘球菌门极显著正相关(P<0.01);nirK基因与拟杆菌门极显著正相关(P<0.01);nirS基因与绿弯菌门、拟杆菌门和粘球菌门极显著正相关(P<0.01)。
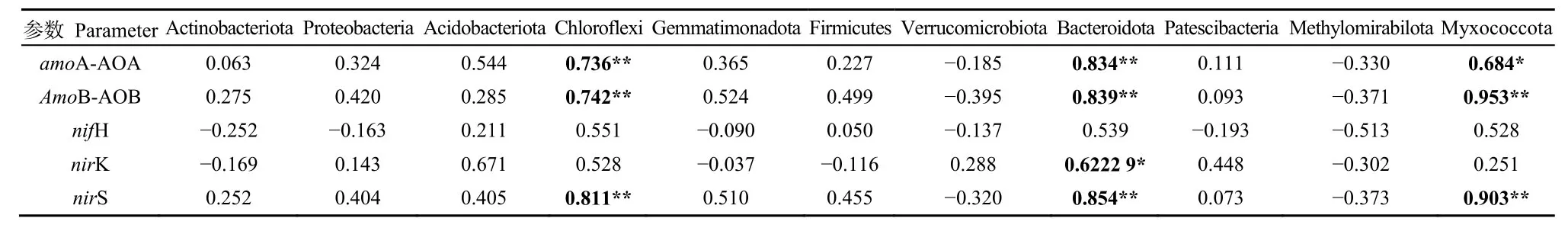
表3 参与氮循环主要过程的功能基因与优势门的相关关系Table 3 Correlation between dominant phylum and functional genes involved in the main processes of nitrogen cycling
3 讨论
3.1 退耕还草过程中植物与土壤性质的变化
我们的结果显示恢复 15年的草地植物生物多样性最高,随着恢复年限的增长,草地优势种发生变化,生物多样性降低。与Zhang et al.(2016)和Wang et al.(2009)的研究一致,植物多样性与丰富度在退耕后呈驼峰型变化,退耕还草后植物多样性与丰富度持续增加,在植物群落演替中期物种丰富度最高,而后随着退耕年限的增加而减少。这是由于恢复初期机会物种先占据,随后随着累积资源的增加,物种丰富度增加。随着生态系统的成熟,强大的竞争对手可能会在后期占据主导地位,导致物种丰富度下降,反映了植物在草地恢复过程中由于环境变化和种间竞争而导致群落结构的变化(海旭莹等,2020)。
退耕初期,因为停止了肥料的输入,土壤养分含量显著降低。然而,随着自然恢复年限的增加,研究发现地上植被发生次生演替,土壤有机残体和根系分泌物的输入量逐渐增加(Zhang et al.,2016),与该研究的结果一致,土壤中积累的养分水平(TC、TN)随退耕年限逐渐增加,且蔗糖酶活性也逐渐增加,说明自然恢复导致了草地碳氮的积累。植被自然恢复过程中,植物的枯枝落叶可将部分养分归还土壤,土壤碳含量积累可能归因于凋落物以及根系的数量和质量逐渐增多,促进了土壤碳的矿化过程(Li et al.,2004);此外,随着恢复年限的增加,与固氮作用有关的豆科植物增加促进了氮的积累(张文彦等,2010);我们的结果均体现出植被自然恢复对土壤系统有积极影响。
3.2 退耕还草过程中细菌群落结构的变化
地上植被演替影响植物根系的生长,引起土壤细菌群落组成和多样性的变化。本研究中,没有额外施肥的退耕草地放线菌门更丰富,这是由于放线菌门对营养物质的贫乏有较高耐受性,且对干燥环境有较高抗性(Schimel et al.,2007)。植被自然恢复过程显著增加了绿弯菌门的丰度,绿弯菌门是环境中C、N等元素生物地球化学循环的重要参与者,且与土壤中积累的养分水平(TC、TN)显著相关。Tscherko et al.(2005)证明,在植被演替期间,土壤C和N资源对确定土壤微生物群落组成非常重要。Lozano et al.(2014)也报告了废弃耕地细菌群落组成沿退耕年限顺序的变化,显示出植被自然恢复对地下微生物群落具有显著作用。
随着长期的植被自然恢复,土壤营养资源不断改善,为微生物提供了更多的可用生态位。关于植被演替对土壤细菌群落多样性和结构的影响的尚有争议(Hossain et al.,2011;Lin et al.,2012)。我们的结果表明,随着恢复时间的推移,细菌群落多样性增加,且土壤细菌群落的α多样性与植物群落的α多样性之间有显著相关关系,表明植物和微生物群落的演替过程是同步的,Zhang et al.(2016)的报道也支持了我们的结果。Lozano et al.(2014)发现了弃耕地土壤植被恢复过程中细菌群落演替阶段之间的明显分离,但与Kuramae et al.(2011)的研究结果相反,他们发现了大量的微生物群落重叠,在草原的时间序列上没有明显的差异。我们基于Bray-Curtis的NMDS结果与Lozano et al.(2014)结果一致,显示细菌群落结构沿时间序列发生了显著变化。
相关分析和RDA表明,优势菌门(放线菌门、变形菌门、酸杆菌门和绿弯菌门)的丰度与土壤养分水平显著相关。土壤总碳、总氮、碳氮比与铵态氮是影响黄土高原亚高山退耕还草自然恢复土地细菌群落结构的主要环境因素。之前的研究表明,土壤铵态氮含量对土壤中主要细菌类群的相对丰度有正向影响(Yao et al.,2014;Yuan et al.,2014),合适的土壤碳氮比含量是微生物群落生长繁殖的驱动力,我们的结果也证实了这点,支持了土壤碳、氮组分在细菌群落形成中的重要性。变形菌门中包含高比例的特定固氮根瘤菌,研究表明氮肥处理对细菌群落结构的影响因研究地域不同存在差异(Bai et al.,2021)。我们的结果显示恢复后期虽C、N积累,但变形菌门的丰度随着自然恢复的时间序列减少,且丰度与土壤水分负相关,与硝态氮和脲酶活性显著正相关,表明在亚高山干旱环境中变形菌门在氮代谢过程中产生了重要作用。此外,结果显示酸杆菌门的Blastocatellia随着恢复时间显著增加,酸杆菌门属于寡营养性型细菌(王光华等,2020),该发现与Campbell et al.(2010)报道的结果不一致,可能与亚高山特殊的地域环境(如低温)有关。
3.3 退耕还草过程中细菌功能类群的变化
氮是土壤中重要的养分元素,土壤氮循环对土壤健康有重要的影响,土壤微生物在氮循环过程中发挥着重要的作用(贺纪正等,2013)。本研究中,由于农田有额外的氮肥输入,农田土壤比恢复草地土壤中的氮代谢相关的基因更丰富,表明农田土壤中固氮、硝化、反硝化作用较弃耕地土壤更强。氮主要通过生物固定进入陆地生态系统,nifH基因是固氮作用的标志性基因,编码固氮酶铁蛋白组分(Wang et al.,2017;Nie et al.,2019)。在自然恢复 30年达到最高值,表明固氮能力随着植被自然恢复时间的推移而加强,我们的结果显示土壤全氮含量与nifH基因丰度显著相关,这与在西藏高山草地土壤和长白山温带森林土壤中的研究结果一致(Yang et al.,2013;Tang et al.,2018)。amoA-AOA与amoA-AOB基因驱动土壤中氨氧化过程,该过程是硝化作用的第一步(Schmidt et al.,2019)。本研究发现amoA-AOA基因在草甸自然恢复20年达到最高值,amoA-AOB基因随自然恢复显著增加,且与总碳、总氮含量显著正相关,大部分研究结果均认为底物浓度是影响氨氧化微生物在土壤中生长并发挥氨氧化功能的主要因素(Martens et al.,2009),本研究也证实了这一结果。同时,amoA-AOA和amoA-AOB基因与绿弯菌门、拟杆菌门和粘球菌门显著正相关,表明这3个优势门可能参与了氨氧化过程,nirS和nirK基因是将亚硝酸盐还原为NO的功能基因,是反硝化作用的标志性反应(Wang et al.,2020)。本研究中随着草甸自然恢复nirS和nirK基因增加不显著((P>0.05)),表明亚高山草甸的自然恢复没有显著增加土壤的反硝化作用。研究发现土壤含氮量等是微生物反硝化过程的重要影响因素(Blaud et al.,2017;王杨,2014),本研究也发现nirS与总氮、硝态氮和铵态氮含量有显著的相关关系。因此,亚高山退耕还草自然恢复过程不仅会改变微生物群落结构,还会通过微生物功能基因丰度的变化而显著影响到土壤中的物质循环过程(Zhang et al.,2007)。
4 结论
亚高山草甸自然恢复显著改变了地上植被的多样性,土壤的养分条件也随着土壤有机残体的积累显著改善。植被恢复改善了亚高山的土壤养分资源,为微生物提供提了更多生态位,土壤细菌群落的多样性沿自然恢复年限顺序显著增加,群落结构也发生了显著变化。同时,随着地上植被的恢复演替,固氮作用的标志性基因 nifH基因丰度显著增加,参与氨氧化过程的amoA-AOA和amoA-AOB基因丰度也显著增加,表明亚高山草甸自然恢复加强了土壤的固氮与硝化过程。通过对环境因子与细菌群落的结构和功能基因分析证实,土壤碳氮含量是影响亚高山退耕还草自然恢复土壤细菌群落的主要环境因素。本研究全面关联了植被、土壤性质、微生物群落结构与氮循环相关功能基因,为制定亚高山退耕还草措施提供了数据依据,并为亚高山草地生态恢复策略提供了理论支撑。
猜你喜欢
科学技术与工程(2022年26期)2022-11-01
当代水产(2022年8期)2022-09-20
中国音乐学(2022年2期)2022-08-10
中国农学通报(2022年14期)2022-06-01
野生动物学报(2022年2期)2022-05-16
昆明医科大学学报(2022年2期)2022-03-29
科教新报(2020年27期)2020-07-31
学校教育研究(2020年7期)2020-04-09
湖北农业科学(2019年22期)2019-12-23
当代工人(2019年22期)2019-12-20
