
MOFs衍生纳米酶在肿瘤治疗中的研究进展
2022-10-10 14:32覃丽婷徐柏龙刘惠玉
中国材料进展 2022年9期
覃丽婷,孙 芸,徐柏龙,刘惠玉
(北京化工大学 有机无机复合材料国家重点实验室,北京 100029)
1 前 言
癌症是当前世界上严重威胁人类健康的重大疾病,具有难治愈、易复发等特点。临床上常用的癌症治疗手段有手术切除、化疗、放疗等,然而这些传统的癌症治疗手段存在副作用大和患者依从性差等问题[1-3]。此外,肿瘤微环境(tumor microenvironment,TME)的特征,如乏氧、微酸性、高浓度的谷胱甘肽(glutathione,GSH,≈10×10-3mol·L-1)和H2O2((50~100)×10-3mol·L-1)等,会导致恶性实体瘤表现出免疫抑制、抗氧化应激、易增殖和转移,严重影响癌症治疗效果[2, 4-6]。因此,TME调节对消除恶性肿瘤至关重要。近年来,TME响应性纳米酶被开发用于肿瘤治疗。在微酸环境下,纳米酶可通过模拟过氧化物酶(peroxidase,POD)活性将H2O2分解为高毒性的活性氧(reactive oxygen species,ROS),从而诱导癌细胞死亡;利用氧化酶(oxidase,OXD)或谷胱甘肽过氧化物酶(glutathione oxidase,GSHOx)活性可催化氧气(O2)产生H2O2或氧化还原型GSH以降低生物体的损伤;模拟过氧化氢酶(catalase,CAT)可将H2O2转化为O2来缓解肿瘤乏氧[5, 6]。基于此,发展具有内源性响应和肿瘤特异性的纳米酶催化疗法可以有效缓解肿瘤乏氧、放大肿瘤氧化应激、逆转免疫抑制,从而能够特异性地杀死肿瘤细胞,并且不会对周围正常组织产生毒副作用,因而具有广阔的应用前景[7, 8]。
自2007年阎锡蕴课题组发现Fe3O4纳米颗粒可以模拟POD活性以来[9],越来越多的纳米材料诸如金属纳米颗粒、金属氧化物、碳基材料和金属有机框架(metal-organic frameworks, MOFs)等的类酶活性相继被发现[10]。其中,MOFs及其衍生物作为纳米酶,具有催化活性高、比表面积可调和表面易修饰等优势,引起了研究者们的广泛关注[11, 12]。一般来说,MOFs作为前驱体或模板经过热解后可以得到能够表现特定的类酶活性或多种类酶活性(包括类CAT、POD和GSHOx等)的纳米酶。并且其可以保持MOFs固有的多孔性质,有利于电子转移和物质传递,从而提高了纳米酶的催化活性,使得MOFs衍生纳米酶在生物医学领域中显示出巨大的应用前景[13]。本综述系统性地总结了MOFs衍生纳米酶的类型,讨论了MOFs衍生纳米酶催化活性的调控策略,详细回顾了MOFs衍生纳米酶介导的肿瘤治疗的最新进展。最后,提出了当前MOFs衍生纳米酶在生物医学应用中仍面临的挑战和未来展望。
2 MOFs衍生纳米酶的分类
由于具有结构多样和功能可调的特点,MOFs已经被广泛用作构建MOFs衍生纳米酶的牺牲模板和前驱体。目前,已发展的MOFs衍生纳米酶包括金属氧化物、金属/碳化物、金属氧化物/碳、金属硫化物和单原子纳米酶(single-atom nanozymes, SAzymes)等。值得注意的是,这些纳米酶在一定程度上可以继承MOFs大的比表面积,以及多孔、活性位点丰富的结构特点,并表现出优异的催化性能[14],为构建纳米酶开辟了一条新的途径。本节将对MOFs衍生纳米酶的种类进行综述和讨论。
2.1 MOFs衍生的金属氧化物纳米酶
以MOFs为前驱体构建的金属氧化物纳米酶,在能源、催化、传感器等领域得到了广泛探索。通过水/溶剂热处理和离子交换等方法合成MOFs前驱体后,在空气气氛条件下热解,周期性排列的金属离子将直接转化为均匀分布的金属氧化物纳米颗粒,部分碳组分被氧化成气态CO2/CO并从骨架中逸出,同时有机配体转化为多孔碳质结构[14-17]。这类金属氧化物纳米酶具有丰富的氧空位、可变的金属价态和增强的类酶活性。例如,Lv等将ZnCo沸石咪唑酯框架骨架(ZnCo-zeolitic imidazolate framework, ZnCo-ZIF)置于空气中煅烧3 h,制备了多孔双过渡金属氧化物纳米笼(ZnO-Co3O4NCs),该纳米酶表现出优异的类POD活性,在H2O2存在下,可以催化其产生羟基自由基(·OH)并将无色的四甲基联苯胺(TMB)氧化形成蓝色产物,即氧化型TMB(oxTMB)[18]。Chen等采用ZIF-67和[Fe(CN)6]3-阴离子交换反应制备了ZIF-67@Co-Fe普鲁氏蓝类似物(PBA)蛋黄壳多面体,并将其置于空气中煅烧得到了Co3O4@Co-Fe氧化物双层纳米笼(DSNCs)。透射电子显微镜(TEM)照片证明了该氧化物具有双壳结构(图1a),其独特的双壳纳米结构可以同时作为活性位点和底物通道(图1b),使得高类POD活性成为可能(图1c)[19]。最近,Zeng等将牛血清蛋白(BSA)封装在Mn-MOF中,在空气中煅烧后,得到多孔Mn3O4纳米粒子(bMn3O4)。在H2O2存在下,具有双重酶活性(类POD和OXD)的bMn3O4能够增强胆固醇氧化酶(ChOx)的催化活性,使得ChOx-bMn3O4杂交纳米酶可以高灵敏度地检测血清样品中的胆固醇[20]。
2.2 MOFs衍生的金属/碳纳米酶
基于MOFs具有丰富的金属节点和有机配体的特点,将其在惰性气氛(如Ar和N2)下进行热解可以将有机配体转化为多孔碳结构,同时MOFs中的金属阳离子被还原成金属纳米颗粒。通过控制热解条件,可以得到不同碳基质比例和金属纳米颗粒尺寸的金属/碳纳米酶[16, 21, 22]。此外,这样制备得到的碳基体不需要额外的稳定剂就可以作为锚定金属纳米颗粒的载体,避免了由稳定配体引起的金属纳米颗粒表面氧化的问题,同时也防止了金属纳米颗粒的聚集,从而增加纳米酶的比表面积并提高其酶活性[13]。例如,Tan等通过热解Cu基MOFs(HKUST-1)制备了金属/碳纳米复合材料(Cu NPs@C),表面不含稳定剂的Cu NPs在MOFs衍生的碳基质中高度分散,使得Cu NPs@C具有优异的类POD活性,并且其对H2O2的亲和力高于天然辣根过氧化物酶(horseradish peroxidase, HRP)[23]。类似地,Song等将Cu基MOFs(HKUST-1)置于500 ℃和N2气氛下碳化10 h,制备了Cu纳米颗粒嵌入的多孔碳复合材料(Cu@C-500),用于比色葡萄糖传感(图1d)。TEM照片证明,Cu纳米颗粒存在并均匀分散在多孔碳复合材料中(图1e)。在H2O2存在条件下,Cu@C-500的类POD活性高于前驱体HKUST-1(图1f)[24]。2018年,Li等通过在N2气氛下对二甲基咪唑钴(ZIF-67)进行热解,得到了Co和N共掺杂的介孔碳纳米酶(Co,N-HPC)。在热解过程中,Co2+被热还原成均匀的Co NPs,使得Co,N-HPC具有优异的类OXD活性,用于GSH检测[25]。

图1 金属有机框架(MOFs)衍生的金属氧化物纳米酶和金属/碳纳米酶:(a)Co3O4@Co-Fe的TEM照片,(b)Co3O4@Co-Fe的类过氧化物酶(peroxidase, POD)活性示意图,(c)Co3O4@Co-Fe在不同H2O2浓度下催化四甲基联苯胺(TMB)氧化的紫外可见光谱[19];(d)Cu@C-500的合成及催化示意图,(e)Cu@C-500的TEM照片,(f)不同材料的类POD活性的紫外可见光谱,其中,i:Cu@C-500,ii:HKUST-1,iii:铜箔,iv:活性炭[24]Fig.1 Metal-organic frameworks (MOFs)-derived metal oxide and metal/carbon nanozymes: (a) TEM image of Co3O4@Co-Fe, (b) schematic illustration of POD-like activity by Co3O4@Co-Fe, (c) UV-vis absorption spectra of TMB oxidation catalyzed by Co3O4@Co-Fe at different H2O2 concentrations[19]; (d) schematic illustration of synthesis of enzyme-like catalysis of Cu@C-500, (e) TEM image of Cu@C-500, (f) UV-vis absorption spectra of POD-like activity of different materials, i: Cu@C-500, ii: HKUST-1, iii: copper foil, iv: activated carbon[24]
2.3 MOFs衍生的金属氧化物/碳纳米酶
通过在惰性气氛下热解MOFs前驱体、空气中氧化这2个步骤,可以将金属氧化物固定在碳质结构上,从而形成金属氧化物/碳纳米酶,显示出优异的催化性能。例如,Wang等报道了一种Cu掺杂的氧化钴多孔碳纳米复合材料(CuCo(O)@PCNs)。先通过在N2气氛下对Cu/ZIF-8@ZIF-67进行热解,合成Cu/Co锚定N掺杂碳纳米管中空多面体(Cu/Co-NCNHP);之后将Cu/Co-NCNHP置于空气中煅烧,得到了具有类CAT活性的CuCo(O)@PCNs(图2a),可与H2O2反应生成O2(图2b)[26]。类似地,Fan等利用静电吸附作用制备得到具有碳基质前驱体的ZIF-8@GO,并将其先后置于在Ar和空气中进行煅烧,并负载热响应电刷(thermally responsive brushes, TRB)得到了具有优异抗菌活性的TRB-ZnO@G,并且不会对正常皮肤组织造成损伤[27]。最近,Xu等以MnCo普鲁士蓝(MnCo-PBA)为前体,制备了具有蛋黄壳纳米笼形貌的MnCo双金属氧化物碳纳米酶(MnCo@C NCs)。该纳米酶具有包括类OXD、CAT和漆酶在内的多种类酶活性,并且表现出优异的催化性能,可用于抗坏血酸、2,4-二氯苯酚和肾上腺素的快速比色检测[28]。
2.4 MOFs衍生的金属硫化物纳米酶
通过界面工程策略可以对MOFs金属节点和配体网络进行理性设计,其中,引入杂原子和构建异质结构可以促进纳米酶表面的电子转移和再分布,从而得到具有复合结构、多金属价态和丰富活性的纳米酶。例如,通过高温煅烧硫化法可以得到MOFs衍生的金属硫化物。将MOFs前驱体置于N2气氛中并加入硫源一起进行煅烧,在煅烧过程中,金属离子被硫化,MOFs的有机配体被碳化从而原位封装形成金属硫化物[29]。Xiong等采用HKUST-1作为前体、Na2S·9H2O充当硫源,制备了具有高类POD活性的PCuS纳米颗粒,并将其作为一种新型的POD用于比色检测[30]。Wang等将硫粉与Co/Mo-MOF置于N2气氛下进行热解,合成了CoS2/MoS2纳米片(图2c)。为了研究CoS2/MoS2的酶催化机制,选择电子自旋共振(electron spin resonance, ESR)进行了测试。实验表明,当CoS2/MoS2与H2O2混合时,ESR图谱中超氧阴离子(·O2-)信号显著增强,证实了CoS2/MoS2具有类酶活性(图2d)[31]。
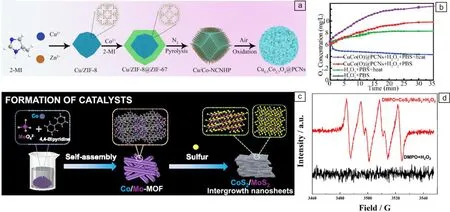
图2 MOFs衍生的金属氧化物/碳和金属硫化物纳米酶:(a)CuCo(O)@PCNs的制备过程示意图,(b)CuCo(O)@PCNs在不同条件下的O2生成曲线[26];(c)CoS2/MoS2纳米片的合成示意图,(d)电子自旋共振(electron spin resonance, ESR)测试·O2-的产生[31]Fig.2 MOFs-derived metal oxide/carbon and metal sulfide nanozymes: (a)schematic illustration of preparation process of CuCo(O)@PCNs, (b) O2 generation curves of CuCo(O)@PCNs under different conditions[26]; (c) schematic illustration of CoS2/MoS2 synthesis, (d) ESR spectra demonstrating ·O2- generation[31]
2.5 MOFs衍生的单原子纳米酶
在催化过程中,纳米酶表面通常仅有少量活性原子起作用,导致活性位点密度低、原子利用率不足和催化机制不明确。受天然金属酶特定空间结构的启发,构建简单配位结构的SAzyme是克服上述缺陷的有效途径。通过调控表面原子结构、电子结构,纳米酶的金属利用效率显著提升,这有助于提升其催化活性。目前,研究者们发展了包括缺陷工程、牺牲模板、电化学腐蚀和高温热解等在内的合成策略,用于制备高活性SAzyme。通过合理设计金属原子结合配位点,不仅能提升SAzyme的金属负载量,还能改善金属前体的原子分散程度。其中,以MOFs为异质原子掺杂的载体,在特定气体(如H2、N2、Ar和NH3)的保护下,通过热解获得具有均匀分布活性位点的SAzyme是一种备受关注的新策略[32]。在热解过程中,MOFs中的金属节点可以原位转化为碳载体上的孤立单原子金属位点。该策略具有易于引入杂原子锚定金属原子和高金属负载的优点,从而可以有效提高纳米酶活性,同时还能通过活性位点的精确识别来阐述构效关系,促进纳米酶的合理设计[13, 33, 34]。在作者研究组以ZIF-8为模板制备了平均直径为130 nm的单原子纳米酶(porphyrin-like metal centers nanoparticles, PMCS)(图3a),通过高角度环形暗场扫描透射电子显微镜(HAADF-STEM)证实了PMCS中含有原子分散的锌原子(图3b),同时,由于其不饱和的Zn-N4活性位点使其具有优异的类POD活性(图3c)[35]。该工作提出的单原子纳米酶的概念极大地推动了纳米酶领域的发展。此外,作者研究组还证明了PMCS在近红外(near infrared, NIR)激光照射下类酶活性进一步增强。负载PMCS的微针系统表现出良好的抗菌性能,实现了光控增强酶活性的伤口管理应用[36]。Huang等在N2气氛下对MOFs封装的铁酞菁进行热解,制备了一种具有高类OXD活性的碳纳米框架轴向N配位的单原子铁纳米酶(FeN5SA/CNF)。通过理论计算和实验表征对比FeN4SA/CNF和MN5SA/CNF(M=Mn,Fe,Co,Ni和Cu)的类OXD活性,发现FeN5SA/CNF具有最强的类OXD活性。这是由于FeN5SA/CNF的单原子活性位点的催化行为类似于细胞色素P450的轴向配位血红素[37]。随后,作者研究组报道了一种三聚氰胺介导的热解活化策略,得到了与HRP结构高度相似的五配位结构的铁基单原子纳米酶(FeN5SAzyme)(图3d)。与不含Fe原子的碳纳米球(monodisperse carbon nanospheres, MCS)相比,FeN5SAzyme表现出更高的类POD活性(图3e)[38]。这些研究表明,SAzyme的催化活性主要取决于其活性中心的空间结构,因此,通过研究不同金属原子活性中心有利于揭露不同的SAzyme的类酶活性和催化机制。基于此,Cao等合成了20种不同类型金属原子N配位中心的人造金属纳米酶(artificial metalloenzymes,AMEs),扫描电子显微镜(SEM)照片显示,制备的AMEs具有相似的纳米立方体形态和粒径(图3f)。通过研究它们的类OXD、POD和卤素过氧化物酶(HPO)的催化活性,揭示不同的活性金属中心对纳米酶催化活性的影响。通过密度泛函理论(density functional theory,DFT)计算发现,AMEs的催化活性与金属中心的电子结构高度相关。由于Fe-AME与H2O2底物的相互作用强于其他AME,且生成·O2-的能垒更低,其表现出最高的类OXD(图3g)和HPO活性,而Cu-AME则表现出最高的类POD活性(图3h)[39]。

图3 MOFs衍生的单原子纳米酶:(a)单原子纳米酶(PMCS)的TEM照片,(b)PMCS的亚埃分辨率的HAADF-STEM照片,部分单个Zn原子用红色圈出,(c)不同浓度的PMCS的类POD活性[35];(d)FeN5 SAzyme的制备过程示意图,(e)不同材料的类POD活性比较[38];(f)人造金属纳米酶(artificial metalloenzymes,AMEs)的SEM照片,(g)不同AMEs的类氧化酶(oxidase, OXD)活性比较,(h)不同AMEs的类POD活性比较[39]Fig.3 MOFs-derived SAzyme: (a) TEM image of PMCS, (b) sub-angstrom resolution HAADF-STEM images of the PMCS, partial single Zn atoms are circled in red, (c) POD-like activity of PMCS at different concentrations[35]; (d) schematic illustration of preparation process of FeN5 SAzyme, (e) comparison of POD-like activity of different materials[38]; (f) SEM images of AMEs, (g) comparison of OXD-like activity of different AMEs, (h) comparison of POD-like activity of different AMEs[39]
3 MOFs衍生纳米酶的活性调控策略
结构和功能对纳米酶的设计至关重要,通过模拟天然酶的催化位点或催化机制可以合理构建MOFs衍生纳米酶,但是如何进一步提高其活性仍是目前研究的热点。近年来,研究者们提出了一些具体的策略来提高酶催化活性,包括表面结构调控、杂原子掺杂、构建双金属MOFs前体和基于MOFs的配体交换策略。
3.1 表面结构调控
研究表明,纳米酶的形态会在很大程度上影响其催化活性,对纳米酶的形态调控主要涉及到形貌、尺寸、比表面积以及孔径。一般来说,尺寸小而孔径大的纳米酶的催化活性要高于尺寸大而孔径小的纳米酶,这是因为增加的比表面积和孔体积可以为反应提供更多的活性位点[40, 41]。Wang等使用不同的软模板(如Pluronic F127和Pluronic P123)或无模板制备了具有不同比表面积的纳米酶(分别记为Co3O4-F、Co3O4-P和Co3O4)。由于Co3O4-F具有更小的尺寸和更多的孔结构,使其比表面积(244.4 m2·g-1)远远大于Co3O4-P(192.6 m2·g-1)和Co3O4(168.2 m2·g-1),故能为催化反应提供更多的活性位点,因此Co3O4-F具有最高的类酶活性[42]。此外,由于不同形态的纳米酶具有不同的暴露面,纳米酶的催化活性还可以通过控制其形态来调节[40, 43]。Song等制备的CuCo2O4纳米棒,由于其大比表面积和多孔结构有利于底物与催化位点接触,使得CuCo2O4表现出优异的类OXD(图4a)和CAT活性(图4b)。与已报道的Co-MOFs衍生的块状Co3O4纳米片相比,介孔CuCo2O4纳米棒对TMB具有更高的亲和力(图4c),这是因为块状纳米片结构缺乏有效的催化位点[44]。多孔碳载体能够为活性金属原子提供丰富的表面积和通道,有利于金属原子催化活性的提升。例如,Niu等将原子分散的Fe掺杂在MOFs衍生的多孔碳中,形成了Fe-NC单原子纳米酶(Fe-NC SAN)。由于单原子Fe位点的充分利用和多孔碳载体的大比表面积,其作为POD模拟物的比活性高达57.76 U·mg-1,可以与HRP相媲美[45]。
3.2 杂原子掺杂
杂原子(如B,P,N,S等)掺杂被认为是一种有前景的活性调控策略。通过杂原子电负性的差异来调节中心金属原子的电子结构,以此改善纳米酶表面的电子性质。此外,杂原子的引入使其与金属原子之间具有协同效应,使得纳米酶的催化活性得以提高[46-48]。Jiao等发现在Fe-N-C单原子纳米酶(FeNC SAzymes)中掺杂B原子可以调节Fe-N4的电子结构,降低反应中间体形成的能垒,从而增强FeNC SAzymes的类POD活性[49]。Ji等构建了一种P和N原子掺杂的单原子纳米酶(FeN3P-SAzyme)。能量色散光谱(EDS)分析表明,Fe,C,N和P元素均匀分散在FeN3P-SAzyme的整个结构中,证明了P和N原子掺杂成功。此外,通过纳米酶催化比色反应的吸收强度对比发现,FeN3P-SAzyme具有优异的类POD活性,其比活性(316 U·mg-1)分别是Fe3O4(9.12 U·mg-1)和无P原子的FeN4-SAzyme(33.8 U·mg-1)的约30倍和10倍。这是因为P和N原子的精确配位可以调节单原子Fe活性中心,并且P原子的电负性弱于N原子,使得FeN3P-SAzyme中的Fe原子的电子转移要少于FeN4-SAzyme,从而增强了FeN3P-SAzyme的催化活性[50]。2020年,该团队通过逆转热烧结过程,将负载在ZIF-8上的Pt纳米颗粒(Pt NPs)直接雾化成单个原子,得到了N,P和S共掺杂的Pt单原子纳米酶(PtTS-SAzyme)。通过对PtTS-SAzyme进行原子结构分析,证明了N,P和S原子的存在(图4d)。此外,为了证明PtTS-SAzyme高的酶活性,将其与用Pt NPs以及不含Pt活性位点的N,P和S掺杂的空心碳多面体(NPS-HC)进行比较。实验证明, P和S原子能够促进Pt NPs向PtTS-SAzyme原子化过程,而且由于P原子的电子捐赠以及N和S原子的电子受体效应,导致单原子Pt催化位点具有独特的电子结构,使得PtTS-SAzyme具有最高的类POD活性(图4e和4f)[51]。
3.3 构建双金属MOFs前驱体
金属元素能够为催化剂提供新的活性位点,在单金属化合物中引入第2个金属位点来调节催化活性是一种提高纳米酶活性的有效策略。与单金属MOFs相比,双金属MOFs的双金属配位结构可以增加催化中心的数量,而且2个金属元素中心的电位差有利于电子转移从而提高催化活性,增强催化能力[52]。通常,在制备原始MOFs的过程中引入二次金属元素,并对该MOFs进行高温煅烧可以将其转化为MOFs衍生的双金属纳米酶。例如,Liu等通过水热法制备了双金属CoMn-MOF-74,并将其置于空气气氛中煅烧6 h,得到了多孔混合双金属氧化物纳米酶(MnCo2O4)。由于过渡金属离子Co(Ⅱ)和Mn(Ⅱ)具有潜在的氧还原活性,可以参加自由基的链式反应,从而加快电子转移速率,使其具有良好的酶模拟活性[53]。Zhang等通过一步水热法构建了Co/Mn氧化物,由于表面不同的氧化价态(如Co2+/Co3+、Mn2+/Mn3+和Mn3+/Mn4+)的氧化还原作用促进了不同价态阳离子之间的电子转移作用,使其比单独的三氧化二锰(Mn2O3)和Co3O4具有更优异的类OXD活性[54]。2020年,该团队还报道了一系列衍生自Co基同源双金属中空纳米笼(HNCs)(C-CoM-HNC, M=Ni,Mn,Cu,Zn)的策略。在该策略中,不仅MOFs前体的结构以可控的方式被改变,而且二次金属离子的引入可以与固有的Co2+形成协同活性位点,使得C-CoM-HNC表现出了比单金属C-Co-HNC更高的类OXD活性[55]。Mu等研究了不同物质的量比例的Fe和Ni的含量对双金属MOFs(FexNiy-MOF)的类POD活性影响,发现2种金属同时存在时其类酶活性要显著高于单金属MOFs(图4g)。通过循环伏安法和ESR表征研究了FexNiy-MOF的催化机理。在H2O2存在下,比较了Fe-MOF、Ni-MOF和Fe3Ni-MOF的还原电流强度顺序,发现它们的还原电流强度顺序与其类酶活性顺序一致(图4h)。这是由于Ni原子的引入提高了FexNiy-MOF的氧化还原能力,加速了TMB和H2O2之间的电子转移,从而提高了Fe3+和Fe2+之间的转化效率,促进了·OH的生成(图4i)[56]。

图4 MOFs衍生纳米酶的活性的调控策略:(a)不同底物溶液的紫外可见吸收光谱(1:ABTS,2:TMB,3:CuCo2O4+ABTS,4:CuCo2O4+TMB,插图为不同底物的溶液颜色变化),(b)不同底物溶液中H2O2分解成氧气的时间依赖性引起的压力变化(1:H2O2,2:H2O2+CuCo2O4,插图为气泡产生的照片),(c)不同材料的类OXD活性比较[44];(d)PtTS-SAzyme的原子结构分析,(e)不同类型的纳米酶在652 nm处的紫外可见吸收曲线,(f)图4e左下角虚线框部分的放大图[51];(g)FexNiy-MOF中不同物质的量比例的Fe和Ni含量的类POD活性,(h)循环伏安法测定不同材料的电流强度,(i)ESR测试不同材料的·OH产生[56]Fig.4 Activity regulation strategies of MOFs-derived nanozymes: (a) UV-vis absorption spectra of different substrate solutions (1: ABTS, 2: TMB, 3: CuCo2O4+ABTS, 4: CuCo2O4+TMB, inset shows color changes with different substrate solutions), (b) time-dependent decomposition of H2O2 into oxygen caused by pressure changes at different substrate solutions (1: H2O2, 2: H2O2+CuCo2O4, inset shows the photo of bubble production), (c) comparison of OXD-like activity of different materials[44]; (d) atomic structure analysis of PtTS-SAzyme, (e) UV-vis absorption curves of different types of nanozymes at 652 nm, (f) enlarge the part of dotted box in the lower left corner of figure (e)[51]; (g) POD-like activity of different molar ratios of Fe and Ni contents in FexNiy-MOF, (h) cyclic voltammetry was used to measure the reduction current intensity of different materials, (i) ESR spectra demonstrating ·OH generation of different materials[56]
3.4 基于MOFs的配体交换
MOFs具有多种有机连接剂和原子分散的金属结构单元,因此,通过对金属节点和有机配体进行合理设计,可以有效调节MOFs的类酶活性[13]。例如,将官能团(—NH2、—NO2等)引入MOFs并且取代其配体上的H原子就是其中一种调控策略,其不仅可以影响金属原子周围的电子云密度,还可以改变催化剂的氧化还原电位和稳定性,使MOFs的微观结构和电子结构得到很好的调控,从而提高酶活性。Wu等选择了与金属蛋白酶具有类似金属-有机配体配位结构的MOFs材料,通过引入—F、—Br、—NH2、—CH3和—OH基团来取代1,4-苯二甲酸(BDC)配体中的H,以此调整BDC配体的电子特性,从而调控MIL-47(V)-X的类酶活性,并将其用于体内抗炎治疗[57]。同时,该团队还采用有机框架MIL-53(Fe)-X纳米酶作为研究模型,通过改变配体的取代基X(X=NH2、CH3、H、OH、F、Cl、Br和NO2)来调整类OXD活性,发现MIL-53(Fe)-X的类OXD活性与配体推拉电子性能之间存在Hammett线性构效关系。通过DFT进一步计算了MIL-53(Fe)-X的结构和能量,揭示了MIL-53(Fe)-X的类OXD催化反应过程中电子传递是整个催化过程的决速步骤,而NO2取代的MIL-53(Fe)-X由于其电子传递步骤中能垒最低,因此其类酶活性最高,是未取代MIL-53(Fe)-H纳米酶活性的10倍[58]。此外,Xu等采用DFT计算来说明—NO2的几何效应和电子效应,进一步从电子结构的角度揭示NO2-MIL-101具有优异催化活性的机理,最后确定NO2-MIL-101活性的增强可能是Fe活性位点的悬键(dZ2)方向上电子减少所致[59]。
4 MOFs衍生纳米酶催化用于癌症协同治疗
近年来,由于催化活性高、底物特异性强和副作用小的优势,MOFs衍生纳米酶已被广泛用于肿瘤治疗领域。通过催化TME中过量的H2O2产生ROS来有效诱导肿瘤细胞死亡,使得MOFs衍生纳米酶在介导催化治疗及其协同疗法(如光动力治疗(photodynamic therapy, PDT)、光热治疗(photothermal therapy, PTT)和化疗等)中取得了一定的进展。本节将对MOFs衍生纳米酶在肿瘤治疗应用中的进展进行综述和讨论。
4.1 MOFs衍生纳米酶介导的催化疗法
近年来,基于纳米酶介导的催化肿瘤治疗引起了广泛关注。然而TME中高水平的GSH往往限制其治疗效果。基于此,Wang等采用一步自组装法合成了具有类POD和GSHOx级联催化活性的六氰铁酸铜(Cu-HCF),通过消耗细胞内的GSH将CuⅡ转为CuⅠ,随后进行类芬顿反应放大类POD活性,从而产生大量的·OH用于诱导肿瘤细胞凋亡[60]。为了提高肿瘤内H2O2的含量,Sang等制备了具有类超氧化物歧化酶(superoxide dismutase,SOD)活性的仿生纳米酶(PZIF67-AT)。其类SOD活性可将·O2-转化为H2O2,从而促进H2O2的产生。此外,负载的3-氨基-1,2,4-三唑(3-AT)可以抑制纳米酶的类CAT活性,在抑制H2O2消除的同时也提高了细胞内的H2O2水平,并能通过类芬顿反应将H2O2转化为·OH,对肿瘤细胞显示出浓度依赖性的毒性(图5a和图5b),并且在动物水平治疗上也抑制了肿瘤生长(图5c)[61]。最近,Cao等以ZIF-8为前驱体,制备了氮掺杂铁卟啉中心的单原子纳米酶(Fe/PMCS)。得益于Fe/PMCS高的类OXD和POD活性以及GSH消耗能力,Fe/PMCS可以选择性地释放大量ROS并剥夺肿瘤中的GSH(图5d)。此外,通过在Fe/PMCS上修饰DNA(mDNA、aDNA和cDNA)可以提高H2O2和催化底物的亲和力,进一步提高ROS的生成量。更重要的是,mDNA可以增强对癌细胞的亲和力,使得macDNA-Fe/PMCS可以特异性地促进癌细胞铁死亡(图5e),表现出了显著的体内治疗效果(图5f)[62]。
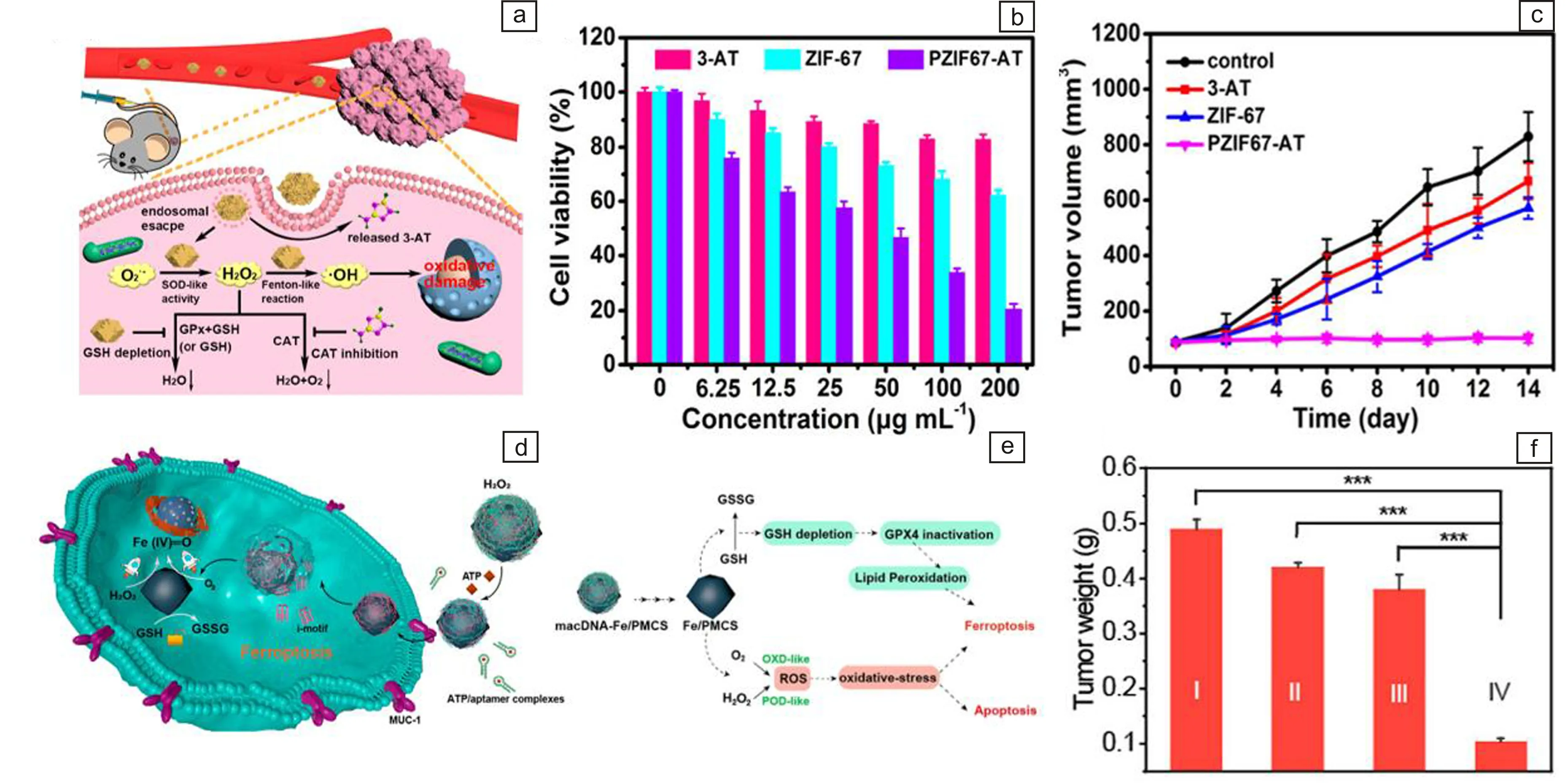
图5 MOFs衍生纳米酶介导的肿瘤催化治疗:(a)PZIF67-AT纳米酶用于肿瘤治疗示意图,(b)不同材料处理后的肿瘤细胞的细胞活力,(c)不同材料治疗后的小鼠肿瘤生长曲线[61];(d)macDNA-Fe/PMCS用于肿瘤治疗的示意图,(e)macDNA-Fe/PMCS通过类酶活性和GSH消耗特异性增强铁死亡的示意图,(f)不同材料治疗14 d后的肿瘤重量,Ⅰ:对照组,Ⅱ:Fe3O4,Ⅲ:macDNA-Fe/PMCS +liproxstatin-1,Ⅳ:macDNA-Fe/PMCS[62]Fig.5 MOFs-derived nanozyme-mediated tumor catalytic therapy: (a)schematic diagram of PZIF67-AT nanozyme for tumor therapy, (b) cell viability of tumor cells treated with different materials, (c) tumor growth curves of mice after different treatments[61]; (d) schematic diagram of macDNA-Fe/PMCS SAzymes for tumor therapy, (e) schematic of macDNA-Fe/PMCS SAzymes enhancing ferroptosis through enzyme-like activity and GSH depletion specificity, (f) weight of the tumor after 14 days treated with different materials, Ⅰ: Control, Ⅱ: Fe3O4, Ⅲ: macDNA-Fe/PMCS +liproxstatin-1, Ⅳ: macDNA-Fe/PMCS [62]
4.2 MOFs衍生纳米酶协同光疗
肿瘤的光疗包括PDT和PTT,其中,PDT是利用光敏剂在激光照射下产生高毒性的ROS,从而诱导细胞凋亡[63]。然而,肿瘤中的乏氧环境极大地限制了ROS水平,显著降低了PDT治疗效果。基于此,Han等制备了MnFe2O4/C纳米酶,通过催化H2O2产生O2以缓解肿瘤乏氧,从而提高PDT的治疗效果[64]。Wang等设计了一种具有类CAT活性的多功能介孔纳米酶(MCOPP-Ce6),可以催化内源性H2O2产生O2,用于缓解肿瘤乏氧。同时,负载的Ce6在NIR激光照射下将O2转化为单线态氧(1O2),杀伤肿瘤细胞(图6a)。为了验证MCOPP-Ce6缓解肿瘤乏氧的作用,在常氧和乏氧条件下进行了细胞死活染色,实验发现在常氧和乏氧条件下观察到相当的红色,说明MCOPP-Ce6可以缓解乏氧并增强PDT的治疗效果(图6b)。此外,肿瘤切片的苏木精和伊红(H&E)染色分析显示,用MCOPP-Ce6加671 nm激光照射处理的小鼠观察到明显的肿瘤组织损伤,证明了纳米酶具有协同PDT治疗的效应(图6c)[65]。为了同时缓解肿瘤乏氧、降低GSH水平,Zeng等设计了MOFs衍生的Mn3O4-PEG@C&A纳米酶,其在缓解乏氧的同时消耗细胞内GSH,并且在NIR激光照射下实现了PDT,有效抑制了小鼠肿瘤的生长[66]。
与天然酶类似,纳米酶也存在最优的反应温度区间,但由于纳米结构的稳定性,纳米酶在较高温度下也能保持高催化活性。因此,通过PTT过程中的光热升温效应可将纳米酶催化反应调整至最佳区间,提升纳米酶催化活性,进而促进ROS产生并高效杀死癌细胞[67, 68]。例如,Liu等测量了NIR照射下TiN NPs的类POD活性,证明了NIR照射引起的热效应可以增强TiN NPs的催化活性[69]。2021年,Zhu等构建了具有优异NIR光热性能的锰基单原子纳米酶(Mn/PSAE),其可以利用H2O2和O2的平行催化反应,同时生成·OH和·O2-,对肿瘤细胞显示出浓度依赖的毒性作用。特别地,在细胞水平实验中,在NIR照射下,Mn/PSAE几乎完全杀死了癌细胞,使得光热协同催化治疗成为可能(图6d和6e)。此外,Zhu等在4T1荷瘤BALB/c小鼠上评价了Mn/PSAE在肿瘤中的光热作用,发现在600 s内小鼠肿瘤部位温度可高达51.9 ℃,并且温度光热转换效率(photothermal conversion efficiency, PCE)达到23.1%,明显高于商用光热试剂IR1048-MZ(20.2%)和吲哚菁绿(ICG,3.1%),更重要的是,Mn/PSAE的酶和光热活性协同治疗使得小鼠模型的肿瘤完全根除(图6f)[70]。考虑到PTT过高的温度在杀死肿瘤的同时会损伤周围正常的细胞组织。因此,Chang等首次提出了一种基于SAzyme的PTT增强铁死亡的新策略。在H2O2和GSH过表达的TME中,具有类POD和GSHOx活性的Pd单原子纳米酶(Pd SAzyme)能够高效催化产生ROS和消耗GSH,导致癌细胞中累积大量脂质过氧化物并引起铁死亡。此外,通过对比不同温度下的模拟酶活性,发现Pd SAzyme在42 ℃下的类POD和GSHOx活性明显优于37 ℃,可用于肿瘤的光热-纳米酶治疗[71]。同样,Zhu等最近报道的新型多功能铜钴纳米粒子(CuCoS NPs)不仅具有优异的光热效应,相比于在室温下的酶活性,其50 ℃下的酶反应速率增加了3倍以上,表明CuCoS NPs具有温度增强·OH的生成能力。此外,CuCoS NPs的类GSHOx活性还能减少·OH的消耗,显示出了显著的抗肿瘤作用[72]。
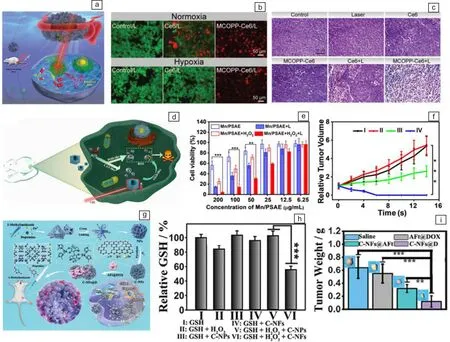
图6 MOFs衍生纳米酶用于肿瘤协同治疗:(a)MOFs衍生的多功能介孔纳米酶协同光动力治疗的示意图,(b)在常氧和乏氧条件下Ce6和MCOPP-Ce6处理后的死活细胞分析,(c)小鼠肿瘤组织的H&E染色[65];(d)锰基单原子纳米酶用于协同光热治疗的示意图,(e)在有无过氧化氢存在下,用Mn/PSAE处理后肿瘤细胞的细胞活力,(f)在有无激光存在的情况下,Mn/PSAE治疗后的小鼠相对肿瘤体积:Ⅰ:PBS,Ⅱ:PBS+NIR,Ⅲ:Mn/PSAE,Ⅳ:Mn/PSAE+NIR[70];(g)C-NFs@D纳米酶协同化疗的示意图,(h)不同处理后对GSH的影响,(i)不同治疗14 d后的平均肿瘤重量[77]Fig.6 MOFs-derived nanozymes for tumor synergy therapy: (a) schematic diagram of MOFs-derived multifunctional mesoporous nanozyme combined with photodynamic therapy, (b) live/dead cell assay for cells treated with Ce6 and MCOPP-Ce6 under normoxic and hypoxic conditions, (c) H&E staining of mice tumor tissue[65]; (d) schematic diagram of Mn-based single-atom nanozyme combined with photothermal therapy, (e) cell viability of tumor cells treated with Mn/PSAE in the presence or absence of H2O2,(f) relative tumor volume in mice treated with Mn/PSAE in the presence or absence of laser, Ⅰ: PBS, Ⅱ: PBS+NIR, Ⅲ: Mn/PSAE, Ⅳ: Mn/PSAE+NIR[70]; (g) schematic diagram of synergistic C-NFs@D nanozymes combined with chemotherapy, (h) effects of different treatments on GSH, (i) average tumor weights after 14 days of different treatments[77]
考虑到NIR触发的PTT与PDT联合协同治疗具有“1+1>2”的治疗效果,Yang等利用原位还原法将超小Pt纳米酶固定在MOFs衍生的碳纳米酶中,用于小鼠结肠癌的PDT/PTT协同治疗,抑瘤率超过90%[73]。Jiang等将光敏剂ICG负载在ZIF-67衍生的中空结构的硫化钴(Co3S4)中,成功制备了具有PDT/PTT效应的Co3S4-ICG纳米酶。在酸性TME中,Co3S4-ICG降解产生的Co2+通过类芬顿反应产生高毒性的·OH,用于肿瘤治疗。正如预期的结果,用NIR激光照射后,Co3S4-ICG组的小鼠肿瘤部位温度在5 min内升高了15.5 ℃,而对照组温度没有显著的变化,说明Co3S4-ICG可以产生足够的热量来消融肿瘤。此外,动物实验证明,当用NIR激光照射时,Co3S4-ICG处理的小鼠肿瘤生长持续受到抑制,这是PDT/PTT协同治疗的效果[74]。
4.3 MOFs衍生纳米酶协同化疗
迄今为止,化疗仍是临床上常用的癌症治疗手段,但其具有严重的副作用。为了改善化疗的毒副作用,Cao等以Ce-MOF作为前驱体进行煅烧,制备了均匀分散的超小多孔CeO2纳米酶(n-CeO2),同时用于负载阿霉素(DOX)。由于出色的类OXD活性和ATP剥夺能力,n-CeO2可以增强癌细胞的氧化损伤、减少肿瘤的能量供应,实现癌症的协同治疗的同时将DOX的副作用降至最低[75]。为了提高肿瘤靶向性、增强化疗效果,Liu等在负载了DOX的单原子铁纳米酶(SAF)上包覆癌细胞膜,有效地将该纳米酶靶向到肿瘤部位,提高了肿瘤抑制效率[76]。2021年,Xing等制备了一种具有独特花状结构的MOFs衍生的纳米酶(C-NFs),并负载DOX(C-NFs@D)用于治疗耐药性肿瘤(图6g)。使用耐药性乳腺癌细胞(MCF-7ADR)作为耐药模型,用于评估体内外的肿瘤治疗效果。同时,由于C-NFs具有类POD活性,可以在TME中催化H2O2产生·OH从而诱导有效的氧化应激,降低GSH水平(图6h)和提高caspase 3的表达以及增强线粒体的有效损伤,进而使细胞对化疗敏感。如预期的结果,C-NFs@D组显著抑制了肿瘤生长(图6i),抑瘤率高达85.4%,实现了较好的协同治疗的效果[77]。
5 结 语
金属有机框架(metal-organic frameworks, MOFs)衍生纳米酶因其丰富的活性位点、良好的孔结构和组成的多样性而备受关注。其中,通过金属节点和有机配体进行直接碳化、氧化和合成后处理,得到的衍生物可能表现出特定的或者多种酶活性,促使MOFs衍生纳米酶在众多用于癌症治疗的纳米酶中脱颖而出。值得注意的是,虽然近年来MOFs衍生纳米酶在生物医学领域取得了一定的成果,但其仍面临着诸多挑战:
(1)通过对MOFs的金属节点和有机配体进行合理设计,可得到具有优异催化活性的MOFs衍生纳米酶。然而,目前用于制备MOFs衍生纳米酶的前驱体主要还是以沸石咪唑酯骨架(zeolitic imidazolate frameworks,ZIFs)系列的材料为主,以其他类型的MOFs为前驱体制备的MOFs衍生纳米酶的研究仍较少,因此,开发其它类型的MOFs用于制备纳米酶有助于更好地模拟天然酶。
(2)与传统纳米酶相比,MOFs衍生纳米酶具有更高的酶模拟催化活性,但煅烧会导致MOFs颗粒的孔径和体积变小、空间位阻变大,使得大分子底物无法进入孔内参与催化反应,导致MOFs衍生纳米酶活性仍低于天然酶。此外,研究表明,分子、纳米粒子和细胞可以被MOFs矿化,导致分子倾向于在孔隙和框架中随机聚集,从而有利于形成大孔隙的MOFs衍生物。因此,可以采用对MOFs进行矿化,或者更先进的热解策略(如用于快速加热的激光和等离子技术)的热解策略,设计出具有更高催化活性的纳米酶。
(3)纳米酶具有成本低、合成方便、可重复使用性好等优点,然而如何提高MOFs衍生纳米酶的化学和生理稳定性仍是癌症治疗中的关键问题。在天然金属蛋白酶中,微环境对稳定蛋白构象和活性位点的微环境对纳米酶的催化稳定性至关重要,因此,可尝试利用氨基酸、多肽或其他生物材料对MOFs衍生纳米酶进行修饰,以模拟天然金属蛋白酶微环境,进而提升治疗过程中的纳米酶催化稳定性。
(4)尽管MOFs衍生纳米酶在癌症治疗方面得到了长足的进展,但其仍存在免疫原性、临床毒性和药代动力学差等问题。因此,对MOFs衍生纳米酶在给药后的药代动力学、生物降解和生理指标进行系统研究和实时监测,进而评估长期毒性以便进一步临床转化是未来需要关注的重点。
猜你喜欢
分子催化(2022年1期)2022-11-02
中国农业科学(2022年16期)2022-09-19
分子催化(2022年3期)2022-08-13
作物学报(2022年3期)2022-01-22
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
电脑知识与技术(2018年19期)2018-11-01
上海医药(2018年15期)2018-09-03
科技创新导报(2016年30期)2017-03-15
