
聚(异丙基甲基丙烯酰胺)@聚(N-异丙基丙烯酰胺)中空微凝胶合成过程的形态学表征
2022-10-08 12:12郭宏博胡西学
分析测试技术与仪器 2022年3期
郭宏博 ,白 露 ,胡西学 ,孙 迪
(1.国家纳米科学中心 技术发展部,北京 100190;2.中国科学院 理化技术研究所,中国科学院光化学转换与功能材料重点实验室,北京 100190;3.国家纳米科学中心 纳米生物效应与安全性重点实验室,北京 100190;4.亚琛工业大学 物理化学,德国 亚琛 52074)
近年来,具备中空核壳结构的聚合物胶体在工业转化和生物医学领域应用广泛,如:催化,敏感微凝胶胶囊,药物输送、诊断、组织工程,“智能”光学系统,生物传感器,微机电系统,涂料和纺织品等行业[1-6].核壳结构可将两种材料结合成一种微球,同时保持二者独有的特性[7-12].因此,核壳结构的微球可以对外部刺激具备多重敏感性,比如pH、温度等.中空核壳多重系统则更有研究意义,外层结构可用来控制调节微球形态,内部空间可来封装材料,二者结合可提供潜在的不同的作用.
聚(N-异丙基丙烯酰胺)(PNIPAM)和聚(异丙基甲基丙烯酰胺)(PNIPMAM)是两种研究广泛的智能温敏聚合物,其在32 ℃(PNIPAM)和42 ℃(PNIPMAM)的水中表现出体积相变(VPT)[13].Berndt等[8,14]利用PNIPAM和PNIPMAM制备出具有核壳结构的微凝胶,发现该类材料可以具有独特的双重温敏特性.Dubbert等[15]将PNIPAM和PNIPMAM应用在微凝胶中,逐步形成双层高分子外壳并将SiO2实心核包裹在内,制备成一种具有同心核壳结构的微凝胶,即PNIPMAM@PNIPAM@SiO2.这类微凝胶同时具备SiO2无机物与上述两种有机物的多重性质[16-17],例如SiO2的高折射率与PNIPAM、PNIPMAM的温度响应特性,由此可用于光子材料等方面的研究[18-19].采用特定的化学试剂将SiO2实心核消除后可得到中空双壳结构的微凝胶即PNIPMAM@PNIPAM[20-22].这种纳米材料具有优异的生物相容性、较大比表面积以及智能敏感的热响应特性,能够在药物递送与释放、存储与保护方面发挥优越的性能[23-27].
目前,在PNIPMAM@PNIPAM中空双壳微凝胶相关报道中,应用较多的表征方法有动态光散射(DLS)、透射电子显微镜(TEM)、原子力显微镜(AFM)等.而结合扫描电子显微镜(SEM)与TEM表征其独特三维立体以及内部结构的形态学研究较少.与其他检测手段相比,SEM与TEM能够为多步骤、多层次纳米材料的复杂合成过程提供最简捷、直观、精确的表征效果.通过观察不同合成产物的三维立体及内部微观结构,测量不同合成产物的粒径大小,能够高效准确判断试验方法的可行性及有效性,为该类纳米材料的后续研究提供充分的理论依据,具有一定的参考价值.
本文选取中空双壳结构的微凝胶即PNIPMAM@PNIPAM为 研 究 对 象,结 合SEM与TEM观察在其合成过程中的产物PNIPAM@SiO2、PNIPAM@PNIPAM@SiO2及 PNIPMAM@PNIPAM微凝胶的三维立体及内部结构,测量其不同核壳结构微凝胶的粒径大小,并用X射线能量色散光谱(EDS)分析其元素组成加以验证,以阐明PNIPMAM@PNIPAM微凝胶的复杂合成过程,如图1所示.
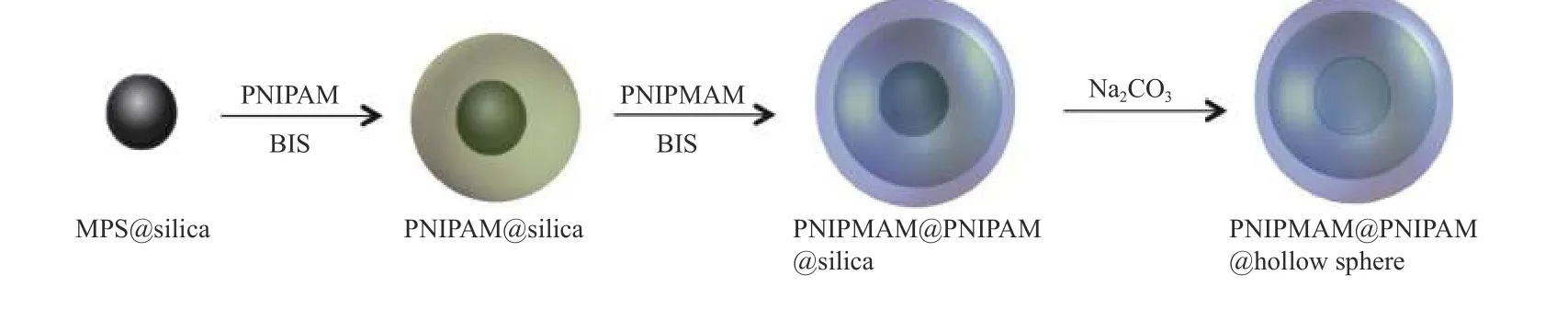
图1 PNIPMAM@PNIPAM中空双壳微凝胶合成过程示意图Fig.1 Synthesis process of PNIPMAM@PNIPAM hollow double-shell microgel
1 试验部分
1.1 仪器与试剂
扫描电子显微镜,SU8200,Hitachi,日本;扫描电子显微镜(SEM)-能谱(EDS),EX-370,Horiba,日本;120 kV透射电子显微镜,HT7700,Hitachi,日本.
氨溶液(Sigma Aldrich);双丙烯酰胺(BIS,Applichem);乙醇(Aladdin);N-异丙基丙烯酰胺(NIPAM,Sigma Aldrich);N-异丙基甲基丙烯酰胺(NIPMAM,Sigma Aldrich);3-(三甲氧基甲硅烷基)丙基甲基丙烯酸酯(MPS,Sigma-Aldrich);过二硫酸钾(KPS,Aladdin);十二烷基硫酸钠(SDS,Macklin);碳酸钠溶液(Na2CO3,Aladdin);聚乙烯吡咯烷酮(PVP,Merck);直径200 nm二氧化硅颗粒(Aladdin,200 nm).上述材料均未经纯化直接使用.
1.2 PNIPAM@SiO2微凝胶合成
PNIPAM@SiO2微球的核壳结构通过Richtering法[15]合成.将1 075 mg NIPAM单体、78.8 mg BIS和100 mg稳定剂PVP溶于100 mL双蒸水中,混合均匀后在70 ℃下使用氮气脱气1 h.加入适量的粒径为200 nm的SiO2纳米粒子,45 min后加入1 mL KPS溶液(10 mg/mL) ,引发聚合反应.10 min后,略微呈乳白色的反应介质变得浑浊,继续在70 ℃下聚合4 h.然后将反应液冷却到室温并搅拌过夜,转速为200 r/min.采用离心法从未反应的单体和短链低聚物中纯化颗粒.
1.3 PNIPMAM@PNIPAM@SiO2微凝胶合成
PNIPMAM外壳的合成方法与上述类似.将515 mg NIPMAM单体、22 mg BIS和1 mg/mL稳定剂SDS的混合物溶解在67 mL双蒸水中,充分搅拌均匀后在70 ℃下使用氮气脱气净化1 h.加入200 mg PNIPAM@SiO2微凝胶颗粒,平衡45 min后加入1 mL KPS溶液(10 mg/mL),启动核-壳-壳结构聚合反应.在70 ℃下聚合4 h,然后将聚合物分散体冷却到室温并搅拌过夜,转速为200 r/min.采用离心法对该步骤合成产物进行纯化.
1.4 中空PNIPMAM@PNIPAM 微凝胶合成
将一定体积的PNIPMAM@PNIPAM@SiO2分散在20 mL Na2CO3溶液(1 mol/L)中, 42 ℃下匀速搅拌72 h.随后,采用透析法对该步骤合成产物进行纯化.
1.5 SEM-EDS表征
将SiO2、PNIPAM@SiO2、PNIPMAM@PNIPAM@SiO2以及中空同心双壳PNIPMAM@PNIPAM这4种微凝胶悬浮液按照合适比例稀释.随后用量程为10 μL的移液枪各吸取上述微凝胶溶液5 μL,分别滴加在粘有导电胶的扫描电镜样品台上,做好样品分组标记.静置1 min后将残留液体吸走,放置在干净密闭的容器内,真空干燥1 h后,采用SEM观察以上4种微球三维立体结构,采用SEM-EDS分析其元素组成.
1.6 TEM表征
将SiO2、PNIPAM@SiO2、PNIPMAM@PNIPAM@SiO2以及中空同心双壳PNIPMAM@PNIPAM这4种微凝胶悬浮液按照合适比例稀释.取若干枚74 μm载有碳膜的铜网,正面朝上放置在干净的封口膜上,使用移液枪吸取上述微凝胶悬浮液各10 μL滴加在铜网上,静置1 min后将残留液体吸走,注意勿将枪头触碰铜网,以免将碳膜戳破.然后将载有样品的铜网放在干净密闭干燥器内,待充分干燥后取出.采用120 kV的TEM观察以上4种微球内部结构并测量其相应粒径.
2 结果与讨论
作为实心核的SiO2形貌像如图2所示.由图2可见,SEM下可以看到表面光滑且粒径均一饱满的SiO2颗粒.TEM下可看到其内部实心结构,尺寸大小约为200 nm.SEM与TEM下均未看到SiO2颗粒团聚现象,分散性良好.

图2 SiO2纳米粒子的SEM、TEM及EDS表征结果(a)(b)为SiO2扫描电镜形态学表征图像(Bar=1 μm),(c)(d)为其透射电镜形态学表征图像(Bar=200 nm),(e)~(j)为SiO2 纳米粒子EDS成分分析结果: (e)为分析区域电子形貌像,(f)为各元素能谱图,(g)为各元素原子百分比柱形图,(h)~(j)分别为C、O、Si元素mapping图(Bar=2.5 μm)Fig.2 SEM, TEM and EDS characterization results of SiO2 nanoparticles(a) (b) SEM morphological characterization images of SiO2 nanoparticles, (c) (d) TEM morphological characterization images of nanoparticles, (e)~(j) analysis results of SiO2 EDS composition: (e) electronic image of analysis area, (f) energy spectrum of each element, (g) bar graph of atomic percentage of each element, (h)~(j) mapping images of C, O, and Si elements, respectively
当NIPAM单体聚合形成PNIPAM薄膜后,SEM下可看到SiO2颗粒表面裹有一层均匀透明的“外壳”,不再似之前光滑.视野内有数个PNIPAM@SiO2微球在同一平面排列整齐,空间上无团聚现象[如图3(a)(b)所示].TEM下可观察到SiO2被紧密包裹形成同心核壳结构,外层PNIPAM薄膜厚度均一,PNIPAM@SiO2较之前粒径增大,测量约为240 nm,PNIPAM层厚度约为20 nm[如图3(c)(d)所示].SEM与TEM下均未捕捉到过量残留的NIPAM单体或SiO2颗粒,说明PNIPAM@SiO2微凝胶经离心后,溶液组成均一稳定.
PNIPAM@SiO2微凝胶成功合成后加入NIPMAM单体,继续合成PNIPMAM层外壳.SEM下可看到与PNIPAM@SiO2相比,PNIPMAM@PNIPAM@SiO2外壳较厚,几乎不透明.在高分子外壳影响下,多个微球容易彼此粘附,空间上堆叠较少[如图4(a)(b)所示].TEM下可看到SiO2外围明显增厚,PNIPMAM层与PNIPAM层边界清晰,形成核-壳-壳同心结构微球[如图4(c)(d)所示].经 测 量,PNIPMAM@PNIPAM@SiO2粒径约为290 nm,PNIPMAM-PNIPAM层厚度约为45 nm.另外,SEM与TEM下偶尔可见少量NIPMAM单体,说明经过离心后该微凝胶体系较为均一.
在PNIPMAM@PNIPAM@SiO2微凝胶中加入碳酸钠溶液,实心核SiO2被消溶,合成PNIPMAM@PNIPAM中空同心双壳微凝胶.SEM下可看到塌陷较为严重以及较轻微的PNIPMAM@PNIPAM[如图5(a)(b)所示],说明微球内部形成空腔.TEM下可明显看到该微球内部电子密度大大降低,几乎中空,双层同心外壳结构依然保留完好,核-壳-壳边界清晰,粒径大小约为250 nm,证明SiO2溶解后,PNIPMAM-PNIPAM双层壳出现一定程度收缩与塌陷[如图5(c)(d)所示].
SEM下随机选取视野,使用EDS对上述4种微凝胶进行成分分析.SiO2微凝胶电子表征区域显示其主要组成元素(C、O、Si、N)含量为0,如图2(e)(g)所示.NIPAM单体围绕SiO2形成高分子聚合物后,检测区域C、O、Si、N这4种元素.N元素出现是因为PNIPAM中亚氨基的存在所导致,从而间接说明PNIPAM@SiO2微球已经成功合成.Si、N元素原子比例约为1.60∶1,如图3(e)(g)所示.PNIPMAM@PNIPAM@SiO2与PNIPMAM@PNIPAM显示其主要元素组成同样为C、O、Si、N,不同的是Si、N元素原子比例分别约为0.85∶1、0.59∶1,如 图4(e)(g),图5(e)(g)所 示.与PNIPAM@SiO2相比,PNIPMAM层外壳的形成引入了更多的亚氨基,从而导致PNIPMAM@PNIPAM@SiO2微凝胶中Si、N元素原子比例的降低.与前两者较之,PNIPMAM@PNIPAM中SiO2被溶解,进一步导致Si、N元素原子比例的再次降低,这些数据也再次阐释PNIPMAM@PNIPAM@SiO2与PNIPMAM@PNIPAM的合成过程,而PNIPMAM@PNIPAM中尚有Si元素的存在,原因可能是微凝胶中空部分有残留的SiO2未彻底消除.

图3 PNIPAM@SiO2微凝胶的SEM、TEM及EDS表征结果(a)(b)为PNIPAM@SiO2微凝胶扫描电镜形态学表征图像(Bar=1 μm),(c)(d)为其透射电镜形态学表征图像(Bar=200 nm),(e)~(k)为PNIPAM@SiO2 EDS成分分析结果:(e)为分析区域电子形貌像,(f)为元素能谱图,(g)为原子百分比柱形图,(h)~(k)分别为C、O、Si、N元素mapping图(Bar=2.5 μm)Fig.3 SEM, TEM and EDS characterization results of PNIPAM@SiO2 microgels(a) (b) SEM morphological characterization images of PNIPAM@SiO2 microgels, (c) (d) TEM morphological characterization images of PNIPAM@SiO2 microgels, (e)~(k): results of EDS composition analysis of PNIPAM@SiO2microgels : (e) electronic image of analysis area, (f) energy spectrum of each element, (g) bar graph of atomic percentage of each element, (h)~(k) mapping images of C, O, Si and N, respectively

图4 PNIPMAM@PNIPAM@SiO2微凝胶SEM、TEM及EDS表征结果(a)(b)为PNIPMAM@PNIPAM@SiO2微凝胶扫描电镜形态学表征图像(Bar=1 μm),(c)(d)为其透射电镜形态学表征图像(Bar=200 nm),(d)中红色虚线所指为核-壳-壳结构每层轮廓边界, (e)~(k)为EDS成分分析结果:(e)为分析区域电子形貌像,(f)为各成份元素能谱图,(g)为各元素原子百分比柱形图,(h)~(k)分别为C、O、Si、N元素mapping图(Bar=2.5 μm)Fig.4 SEM, TEM and EDS characterization results of PNIPMAM@PNIPAM@SiO2 microgels(a) (b) SEM morphological characterization images of PNIPMAM@PNIPAM@SiO2 microgels, (c) (d) TEM morphological characterization images of PNIPMAM@PNIPAM@SiO2 microgels, red dotted line refers to outline boundary of each layer of core-shell-shell structure, (e)~(k) results of EDS component analysis: (e) electronic image of analysis area, (f) energy spectrum of each element, (g) bar graph of atomic percentage of each element, (h)~(k) mapping images of C, O, Si and N elements, respectively

图5 PNIPMAM@PNIPAM中空双壳微凝胶SEM、TEM及EDS表征结果(a)(b)为PNIPMAM@PNIPAM中空双壳微凝胶扫描电镜形态学表征图像(Bar=1 μm),(c)(d)为其透射电镜形态学表征图像(Bar=200 nm),(e)~(k)为PNIPMAM@PNIPAM微凝胶EDS成分分析结果:(e)为分析区域电子形貌像,(f)为各元素能谱图,(g)为其原子百分比柱形图,(h)~(k)分别为C、O、Si、N元素mapping图(Bar=2.5 μm)Fig.5 SEM, TEM and EDS characterization results of PNIPMAM@PNIPAM hollow double-shell microgels(a) (b) SEM morphological characterization images of PNIPMAM@PNIPAM hollow double-shell microgels, (c) (d) TEM morphological characterization images of PNIPMAM@PNIPAM hollow double-shell microgels, (e)~(k) results of EDS component analysis: (e) electronic image of analysis area, (f) energy spectrum of each element, (g) bar graph of atomic percentage of each element, (h)~(k) mapping images of C, O, Si, and N elements.respectively
以上4种微凝胶mapping结果显示各元素分布区域轮廓清晰,与被检测样品组分相吻合,如图2~5中的(h)~(k)所示.C元素分布广泛,在微凝胶内外区域均可看到,外围密度较高,可能是导电胶中含有大量C所导致.O元素在前3组主要高密度聚集在微凝胶中,在PNIPMAM@PNIPAM中由于SiO2被消除,O元素含量大大降低,分布也较为广泛,不再似之前集中.Si、N元素分布基本一致,集中在微球及周边相关区域.上述4组样品中元素的精确定位能够为成分的定性与定量分析提供有力依据.
3 结论
(1)PNIPAM、PNIPAM-PNIPMAM能够很好的形成微凝胶体系中的“壳”,将SiO2颗粒紧密包裹形成同心核-壳结构(PNIPAM@SiO2)及同心核-壳-壳结构(PNIPMAM@PNIPAM@SiO2),利用碳酸钠溶液将SiO2核消溶后,PNIPAM-PNIPMAM依然能够保持良好的中空同心双壳结构即PNIPMAM@PNIPAM微凝胶.
(2)实心核SiO2粒径约200 nm,PNIPAM@SiO2微球粒径约为240 nm,PNIPMAM@PNIPAM@SiO2粒径约为290 nm,PNIPMAM@PNIPAM微球粒径约为250 nm,微球粒径逐渐增大说明聚合物高分子外层成功合成,后者减小说明去掉实心核后PNIPAM-PNIPMAM双壳结构出现一定程度收缩与塌陷.
(3)EDS表征结果中Si、N元素的含量变化及原子比例、分析区域元素分布的精准定位间接为PNIPMAM@PNIPAM中空双壳微凝胶的合成过程提供研究佐证.
(4)利用SEM、TEM对PNIPMAM@PNIPAM合成过程产物进行形态学表征,将其三维空间及内部结构可视化,精准测量其合成过程产物的粒径大小.简洁直观阐明其合成方法的科学性及有效性,为该类纳米材料的后续研究提供充分的理论依据,具有一定的参考价值.
猜你喜欢
原子与分子物理学报(2021年2期)2021-03-29
山西能源学院学报(2021年6期)2021-01-21
凿岩机械气动工具(2017年3期)2017-11-22
西安工程大学学报(2016年6期)2017-01-15
材料科学与工程学报(2016年1期)2017-01-15
造船技术(2016年1期)2016-03-18
湖北师范大学学报(自然科学版)(2015年1期)2016-01-10
新疆钢铁(2015年1期)2015-11-07
建筑材料学报(2015年3期)2015-02-28
华东理工大学学报(自然科学版)(2015年5期)2015-02-27
