
尿嘧啶激发态动力学溶剂效应的飞秒瞬态吸收光谱研究*
2022-09-30 05:41沈环华林强魏政荣
物理学报 2022年18期
沈环 华林强 魏政荣
1) (华中农业大学理学院,武汉 430070)
2) (中国科学院精密测量科学与技术创新研究院,波谱与原子分子物理国家重点实验室,武汉 430071)
3) (湖北大学物理与电子科学学院,武汉 430062)
尿嘧啶是构成RNA 链最基本的结构单元之一,其光辐射下的光物理和光化学过程与光致癌变等过程紧密关联,因而受到广泛的关注.本文采用飞秒瞬态吸收光谱技术,研究了在紫外光辐射下,溶剂效应对尿嘧啶激发态超快动力学的影响.实验利用一束264 nm 的激发光将尿嘧啶激发到1(π,π*)态(即S2 态).然后,利用覆盖范围为280—360 nm 的紫外超连续谱作为探测光,实现对尿嘧啶激发态和基态动力学的同步探测.测量发现,尿嘧啶在乙腈溶剂中显现了两个动力学衰减过程,即9.8 ps (在300 nm 处)和>1000 ps,分别对应于振动冷却过程和三重态的衰减过程.这一观察与水溶液中的结果差异很大,因为在水溶液中基本没有测量到三重态的布居.通过对一系列溶液展开测量,发现尿嘧啶的超快动力学过程依赖于溶剂形成氢健能力的大小.越容易形成氢键的溶剂,尿嘧啶越不容易布居到三重态.
1 引言
核酸碱基是DNA 和RNA 的基本组成部分,是核酸分子中对光反应最敏感的部分.对核酸碱基在紫外光辐射下的光物理和光化学过程的理解,是研究DNA 光诱变损伤和光致癌的基础[1-3],特别是光辐射下核酸碱基的超快动力学过程.因此,该方面研究的展开,不仅在基础研究中具有重要的科学意义,还在生命健康方面具有重要应用价值.
尿嘧啶(U)是最简单的天然核酸碱基.一般认为,在溶液中的尿嘧啶受到260 nm 左右的紫外光照射下被泵浦到激发态,随后在激发态经历内转换过程衰变到基态,这一过程大概在皮秒量级.因此,尽管紫外光在自然界中普遍存在,但是在紫外光诱导下处于激发态的核酸碱基仍然能够快速地回到稳定的基态.这一过程能够使DNA 避免光致损伤并保持其遗传信息[4-8].然而,最近新的研究表明,溶剂效应能够对尿嘧啶以及衍生物的超快衰变动力学过程产生很大的影响[9-17].Hare 等[9]利用飞秒瞬态吸收光谱技术研究了1-环己基尿嘧啶(1-CHU)在质子和非质子溶液中的超快衰变动力学过程.研究发现,在质子溶剂中1-环己基尿嘧啶暗态1(n,π*)的非辐射弛豫时间尺度在几十皮秒,而在非质子溶剂中,其弛豫时间尺度为数十纳秒.他们提出假设,认为质子溶剂能够调节明态1(π,π*)和暗态1(n,π*)的能级.当暗态1(n,π*)的能级和三重态3(π,π*)态的能级相近时,这两个态之间就会发生系间交叉过程.Gustavsson 等[10,11]通过飞秒荧光上转换光谱法测量了5-氟尿嘧啶(5-FU)在水、乙腈和甲醇中的激发态动力学.结果表明,溶剂对明态1(π,π*)的衰变动力学过程有显著影响.这主要是与溶剂的氢键有关,而与溶剂的极性和黏度关系不大.在之前的研究中,我们报道了尿嘧啶及其C5,C6 取代衍生物在水和乙腈溶液中的飞秒瞬态吸收光谱研究,发现这些分子被泵浦到明态1(π,π*)之后,平行衰减到基态S0和中间态1(n,π*).并且,C5和C6 取代延缓了明态1(π,π*)到中间态1(n,π*)的衰减速率[12,13].最近,Li 等[14]采用共振拉曼光谱方法结合理论计算研究了6-氮尿嘧啶(6-AU)S2态的超快动力学过程,他们对S2态主要衰减至S1态的结论与我们瞬态吸收光谱测量的结果一致.Li 等的研究还就溶剂效应对尿嘧啶、6-氮尿嘧啶等体系超快动力学的影响展开了讨论.在理论计算方面,Improta 等[15-17]利用可极化连续模型(PCM)研究了尿嘧啶及其衍生物的溶剂效应,在分子尺度上计算了5-FU 和U 在水和乙腈溶剂中超快衰变的溶剂效应.他们认为这种超快衰变过程有极大可能是受到溶剂与溶质分子之间氢键的强烈影响,因为5-FU 的1(π,π*)态和1(n,π*)态之间的锥形交叉过程将使C(4)=O 在乙腈溶剂中的键长拉长[14].Etinski 等[18]通过量子化学计算发现,6-氮杂尿嘧啶从1(n,π*)到三重态T1的超快系间交叉(intersystem crossing,ISC)过程在真空中为125 ps,而在乙腈溶液中为34 ps.他们的研究认为这种衰变寿命的差异主要受溶剂极性的影响.最近,Danillo 等[19]采用CASPT2 方法和cc-pVDZ 基组,计算了1-环己基尿嘧啶在真空以及水溶液中的超快动力学,并对极性质子型溶剂中三重态产率的抑制现象提出了解释.
如上所述,尽管人们对尿嘧啶及其衍生物的衰变动力学过程做了很多研究,但是溶剂效应对其衰变过程的影响仍未得到全面理解.尤其是在紫外光辐射下的基态和三重态的动力学过程,由于瞬态吸收信号常常出现在紫外波段甚至深紫外波段,一直未能被直接观察和深入理解.然而,这一困难可以通过使用延伸到紫外区域的连续性白光作为探测光来克服.在之前的研究中,我们将探测光延伸到300 nm 以下,这使得能够同时研究尿嘧啶及其衍生物的基态和激发态的衰变动力学过程,从而能够全面揭示取代基对尿嘧啶光辐射过程的影响[12,13].本文采用飞秒瞬态吸收光谱技术,在紫外区域对处于激发态的尿嘧啶在不同溶剂中的超快衰变动力学过程进行了研究.系统讨论了溶剂环境对尿嘧啶的瞬态吸收光谱和激发态弛豫动力学过程的影响.研究发现,尿嘧啶三重态的布居动力学与溶剂的特征紧密相关.
2 实验设备和过程
实验用到的尿嘧啶样品,采购于Alfa Aesar公司.实验用到的溶剂采购于申试化工,为高效液相色谱(HPLC)级.所有的化合物在使用前未经过进一步提纯.所有的实验溶液均在实验室中进行重新配制,从而保证实验样品的可靠性.所有测量均在室温下饱和空气条件下进行,样品溶液的浓度为1 mmol/L.
在之前的研究中,我们详细描述过实验设备[12,13].这里进行一个简要的描述.实验中使用的飞秒激光器是掺钛蓝宝石多通放大器,型号为FemtoPower Compact PRO (Femtolasers Produktions GmbH),中心波长在792 nm,脉冲宽度30 fs,脉冲强度0.8 mJ,重复频率5 kHz.激光器输出的基频光束经过分束器被分成两束.一束基频脉冲经过二倍频晶体BBO 被转换成396 nm 的二次谐波,然后和基频脉冲进行合频产生264 nm 的三次谐波(约2 µJ).这一束脉冲(264 nm)用来激发研究样品到它的激发态1(π,π*)态,即S2态.将产生的396 nm的二次谐波的一小部分(约1 µJ)聚焦在CaF2晶体上用来产生探测光和参考光.产生的连续性白光波长可延伸到深紫外区域,波长覆盖范围为250—600 nm 可见光区域.样品放置在1 mm 厚的流动循环样品池中,以保证激光脉冲每次都与新样品相互作用.通过调节泵浦脉冲和探测脉冲之间的时间延迟,使得这两束脉冲在样品上实现时间和空间上的重合.两束激光脉冲均为线性偏振,它们之间的偏振夹角被设置为魔角(约54°),以消除分子取向效应带来的影响(尽管实验中并未发现激光偏振角度对实验结果的明显影响).两个线性CCD 被用来测量探测光和参考光的光谱信号.实验中使用的激光脉冲的波长均由光谱仪直接测量,而光谱仪则通过已知原子谱线进行校准.根据光学克尔效应,校正了作为探测脉冲的连续性白光对时间零点的影响,整个装置的时间分辨率可达到300 fs 左右.实验中,对数据进行5—10 次的重复采集,然后对平均结果进行处理.
3 实验测量结果及分析
3.1 尿嘧啶在乙腈溶剂中的激发态动力学
实验中选择乙腈作为一种典型的非质子溶剂,用来研究尿嘧啶在该类溶剂中的激发态动力学.尿嘧啶在乙腈溶剂中的稳态吸收谱已经有详细的报道[4,6],本次实验没有再次进行测量.实验首先直接测量了在不同的泵浦和探测脉冲的时间延迟下尿嘧啶在乙腈溶剂中的瞬态吸收光谱,结果如图1(a)和图1(b)所示.图1(a)给出了在较小的时间延迟1—19.5 ps 之间的光谱,而图1(b)则给出了在较大的时间延迟59.5—79.5 ps 之间的光谱.从图1(a)可以看出,随着泵浦光的激发,尿嘧啶被瞬间激发到一个激发态上,该激发态有一个很宽的正吸收带,波长范围覆盖280—310 nm,吸收峰值约在300 nm.在时间延迟从1—9.5 ps 之间,该吸收带一直存在,但在时间尺度上展现了一个快速的衰减,衰减寿命约为10 ps,并在吸收峰值上出现了蓝移现象,吸收峰一直往波长更小的方向移动,其光谱变化趋势如图1(a)中的箭头所示.此外可以发现,瞬态吸收光谱信号在长波范围内(如>310 nm)较弱且衰减较快.这些特征和振动冷却的特性十分相似.从图1(b)可以看出,在时间延迟59.5—79.5 ps 之间,瞬态吸收光谱信号并没有明显的变化.在280—340 nm 之间吸收信号仅观察到非常微小的衰减.在大于340 nm 的范围内吸收信号几乎没有变化,并在79.5 ps 时仍然观察到平稳的吸收信号强度,这说明此时尿嘧啶分子依旧处于激发态.
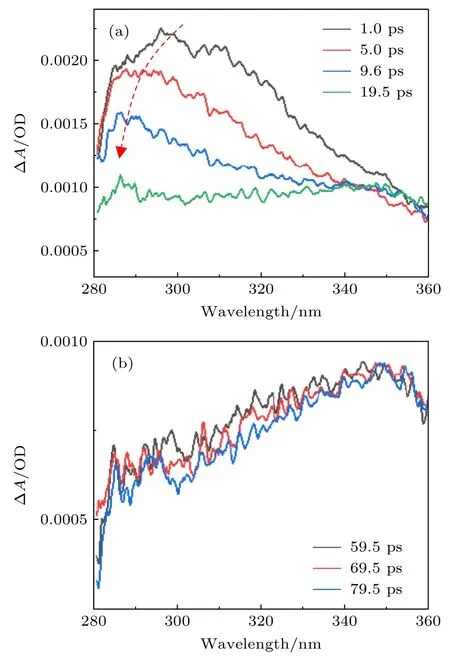
图1 在不同的泵浦和探测脉冲的时间延迟下尿嘧啶在乙腈溶剂中的瞬态吸收光谱 (a) 1—19.5 ps;(b) 59.5—79.5 psFig.1.Transient absorbance change of uracil in acetonitrile at different pump-probe delays: (a) 1—19.5 ps;(b) 59.5—79.5 ps.
为了进一步提取尿嘧啶分子激发态的详细动力学信息,对瞬态吸收光谱中较慢的弛豫过程(>10 ps)进行了全局拟合分析,而对较快的过程在不同波长处进行了寿命的拟合.为了方便展示其主要特征,将300 nm 处的瞬态吸收光谱信号强度随着泵浦-探测脉冲时间延迟的变化曲线,及其最佳拟合曲线均作图于图2 中.从图2 可以看出,除了时间零点附近溶剂产生的背景信号,时间延迟变化曲线由一个快速衰减组分和一个慢速衰减组分组成.经拟合,快速衰减成分的寿命常数为9.8 ps,而慢速衰减成分的寿命常数>1000 ps.这一观察结果与尿嘧啶在水溶剂中的情况差异较大.在水溶剂中观察到的时间延迟曲线也有两个衰减成份,但是衰减寿命常数分别为1.1 和22 ps,且1.1 ps 的快速衰减对应于基态S0的振动冷却过程,而22 ps 则对应于最低的激发态1(n,π*),即S1态的寿命[13].这一结果表明在水溶剂中很难观察到长寿命的系间交叉动力学过程,因为相当大部分的分子在1.1 ps 内就弛豫到了基态.在另一方面,早前的吸收光谱实验报道了水溶剂中尿嘧啶的三重态T1的量子产率大约为0.02—0.04,而在乙腈溶剂中则有0.2[20,21].这意味着在乙腈溶剂中尿嘧啶的三重态T1的量子产率不能被忽略,并且在瞬态吸收光谱中有可能观察到其长寿命的吸收信号.在本次测量中发现,在泵浦-探测时间延迟79.5 ps 之后尿嘧啶分子仍然处于激发态,并且显示了一个非常缓慢的衰减趋势(数据拟合寿命大于1000 ps).因此,我们认为该长寿命的衰减组分可以被标识为其三重态T1的寿命.

图2 在300 nm 处尿嘧啶在乙腈中的瞬态吸收信号在不同的泵浦-探测光时间延迟下的变化曲线Fig.2.Time-dependent absorbance change of uracil in acetonitrile with different pump-probe delays at 300 nm.
在图2 中,除了长寿命的组分,还有一个快速衰减的寿命组分,其时间常数为9.8 ps.这一结果与先前在非水溶剂中的分子基态振动冷却的研究结果类似[22-26].此外,在图1(a)中还观察到了瞬态吸收峰的蓝移现象,这也是基态振动冷却过程中经常观察到的一个典型特征.因此,我们将该组分归因于基态(即S0态)的振动冷却.此外,在探测光波长为295,310,325 和340 nm 处的拟合寿命分别为11,6.9,4.9 和1.7 ps.该寿命随着波长的增加而减少.尽管如此,该寿命仍然要比在类似条件下水溶剂中的寿命要长得多.
本文无法明确分辨最低的激发态1(n,π*)态(即S1态)的吸收.在尿嘧啶分子中,其1(n,π*)态通常被认为是一种过渡态,通过它与T1态的系间交叉过程,可以实现三重激发态的布居[9].这与在水溶剂中不同,在水溶剂中可以清楚地观察到寿命为22 ps 的组分,该组分被认为是1(n,π*)态的寿命[13].然而,我们在乙腈溶剂中并没有清楚地观察到与该寿命相近的组分,但是这不能排除1(n,π*)态被布居的可能性.这可能是因为在乙腈溶剂中尿嘧啶的1(n,π*)态吸收非常地弱,或者它与处于基态的高振动态相叠加,其动力学过程被混合进了基态的振动冷却过程中.
基于以上分析,图3 给出了尿嘧啶在乙腈溶剂中的衰减动力学过程示意图.在264 nm 的激光脉冲激发后,分子被泵浦到1(π,π*)态.处于1(π,π*)的分子通过内转换过程分别同时衰减到1(n,π*)和基态S0的高振动态.随后,一部分分子通过1(n,π*)态衰减到三重态T1态,另一部分处于基态的高振动态的分子则进行振动冷却的弛豫衰减,两个过程对应的时间常数分别为>1000 ps 和9.8 ps.
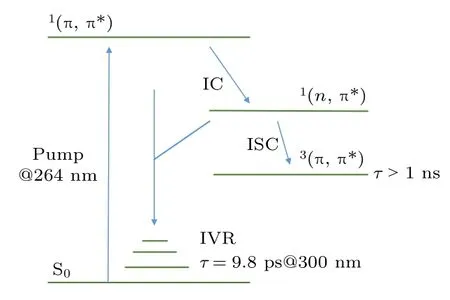
图3 尿嘧啶在乙腈溶剂中的光激发衰减动力学过程示意图Fig.3.Jablonski diagram of the proposed decay mechanism for photo-excited uracil in acetonitrile.
3.2 尿嘧啶在甲醇溶剂中的激发态动力学
与乙腈溶剂不同,甲醇溶剂是典型的质子溶剂,我们也测量了尿嘧啶在甲醇溶剂中的超快激发态动力学.图4(a)和图4(b)给出了在不同的泵浦-探测光延迟下尿嘧啶在甲醇中的瞬态吸收光谱.与乙腈的情况类似,该瞬态吸收信号在280—350 nm区域显示了快速衰减趋势,寿命约为10 ps.此外,在最初的5 ps 内,吸收峰峰值在约290 nm 处,并且也观察到了峰值明显蓝移的现象.同样地,在20 ps 之后吸收光谱在290—350 nm 区域仍然显示出很强的吸收信号,如图4(b)所示.探测光在300 nm 处的瞬态吸收信号随泵浦-探测光延迟的变化曲线也可以被拟合为两个时间常数,即3.5 ps和>1000 ps.由于在乙腈和甲醇溶剂中观察到的瞬态吸收光谱变化规律相似,我们将第一个快速时间常数标识为基态的高振动态的振动冷却过程,而第二个慢速时间常数标识为三重态T1的寿命.
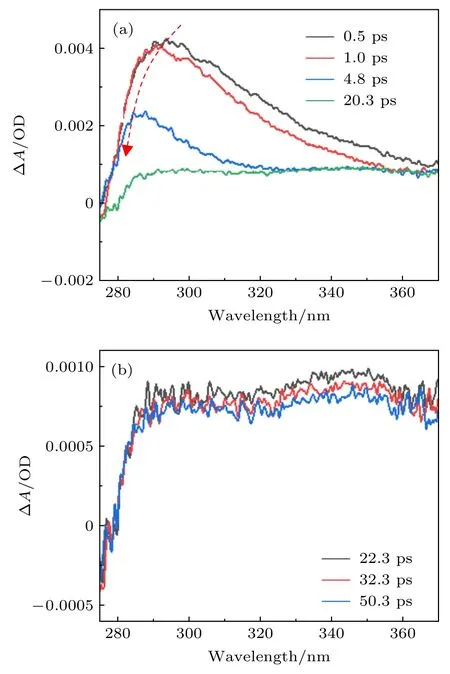
图4 在不同的泵浦和探测脉冲的时间延迟下尿嘧啶在甲醇溶剂中的瞬态吸收光谱 (a) 0.5—20.3 ps;(b) 22.3—50.3 psFig.4.Transient absorbance change of uracil in methanol at different pump-probe delays: (a) 0.5—20.3 ps;(b) 22.3—50.3 ps.
3.3 尿嘧啶激发态动力学的溶剂效应
为了更加系统地研究溶剂对尿嘧啶激发态衰减动力学的影响,我们研究了水、乙腈、甲醇、乙二醇和乙酸乙酯等多种溶剂中的超快动力学(为了论文的可读性,乙二醇和乙酸乙酯溶液的实验数据未详细显示在本文中,水溶液的数据详见参考文献[12,13]).在这些溶剂中,乙腈和乙酸乙酯为非质子溶剂,其他则为质子溶剂,通过比较尿嘧啶在这两类溶剂中的动力学过程可以揭示氢键的作用.此外,这些溶剂的黏度和极性也不相同.因此,这些溶剂的选择可以更好地帮助我们系统地研究溶剂在尿嘧啶激发态衰减动力学过程中所扮演的角色.
通过比较发现,除了水溶剂之外,在其他大多数溶剂中都可以观察到三重态的布居.如果用三重态衰减的振幅和振动冷却过程来计算三重态的相对产率Y,显示出以下趋势:Y(乙腈) &Y(乙酸乙酯)>Y(甲醇) &Y(乙二醇)>Y(水).这一趋势表明,在氢键较强的溶剂中,尿嘧啶以三重态存在的概率较低.还可以注意到,基态的振动冷却过程的寿命显示出类似的趋势,即τ(乙腈) &τ(乙酸乙酯)>τ(甲醇) &τ(乙二醇)>τ(水),如表1 所列.这一趋势表明,氢键加快了振动冷却过程中的能量耗散,从而影响了三重态的产率.另一方面,还发现溶剂的黏度和极性对尿嘧啶的衰减过程的作用几乎可以忽略不计.本文的研究结果与Hare 等[9]的结论具有一致性,即三重态主要来自1(n,π*)态,并且只有当1(n,π*)态具有多余的振动能量时,才能通过系间交叉过程过渡到三重态.本文的观察结果表明,三重态T1被布居的前几个皮秒内,在非质子溶剂中的振动冷却过程很慢(通常在几个或者几十个皮秒内),这使得更多激发态的分子从热态1(n,π*)过渡到三重态T1.
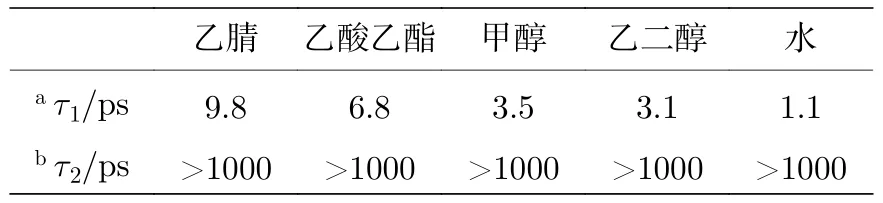
表1 在不同溶剂中观察到的时间常数Table 1.Observed time constants in different solvents.
4 结论
本文利用飞秒瞬态吸收光谱技术,研究了尿嘧啶在不同溶剂中的激发态衰减动力学过程.实验中使用了可延伸至280 nm 的深紫外探测光,更加直接地系统测量了尿嘧啶在不同溶剂中的三重态布居动力学.实验结果为验证三重态布居过程对溶剂的依赖性提供了直接有力的证据.通过对乙腈溶剂中测得的瞬态吸收光谱的详细分析,在300 nm 处直接获得了两个衰减时间常数,即9.8 ps 和>1000 ps.与我们之前在水溶剂中没有观察到明显的三重态的实验结果相比,乙腈溶剂对三重态的布居起到了至关重要的作用.此外,系统测量了不同溶剂中尿嘧啶的瞬态吸收光谱,结果还表明尿嘧啶的衰减动力学对溶剂的氢键存在明显的依赖性.在氢键较强的溶剂中,如甲醇、乙二醇和水溶剂中,尿嘧啶的1(n,π*)态分子转移到三重态的概率较小且分子基态冷却过程较快,这说明氢键加快了内转换过程中的能量耗散速率.
感谢华心仲博士在早期实验数据采集方面的贡献.
猜你喜欢
无线电工程(2022年10期)2022-10-24
昆钢科技(2022年2期)2022-07-08
昆钢科技(2021年4期)2021-11-06
延边大学学报(自然科学版)(2021年1期)2021-04-27
汕头大学学报(自然科学版)(2020年4期)2020-12-14
防爆电机(2020年3期)2020-11-06
分析化学(2018年12期)2018-01-22
北京航空航天大学学报(2017年8期)2017-12-20
北京航空航天大学学报(2017年9期)2017-12-18
山东工业技术(2016年15期)2016-12-01
