
聚乙醇酸材料的加工改性及其水下降解特性的研究进展
2022-09-24 13:22马兰荣尹慧博
中国塑料 2022年9期
马 超,马兰荣,魏 辽,尹慧博,林 祥
(1.北京科技大学化学与生物工程学院,北京 100083;2.中国石化石油工程技术研究院有限公司,北京 102206)
0 前言
石油基聚合物不可生物降解,对微生物降解的抵抗力导致它们被不断储存在环境中,如今塑料废弃物导致的“白色污染”对环境造成了很大的负担。此外,由于石化资源的有限性及全球分布不均,石油价格长期上涨,造成石油基聚合物的价格也不断攀升。随着国家“限塑令”的持续推进,生物基可降解塑料必然成为一种新的应用趋势。预计到2030年,生物可降解塑料的生产将占塑料市场的40%以上。从食品包装、制药、电子、农业到纺织品,生物可降解塑料将在许多应用中替代传统的、不可降解的石化塑料。来自可再生资源的生物基聚合物材料是减少对基于化石燃料的食品包装材料和塑料污染的依赖的理想选择,但许多生物基聚合物材料本身力学性能和使用性能补足,往往需要对其进行改性,从而实现其工业应用价值。PGA作为一种具有突出生物降解性及生物相容性的可再生高分子材料,不管是在自然堆肥的环境下还是在海水中最终都可以降解成无毒无害的二氧化碳和水,并且其优异的生物相容性在人体中也可以降解成对人体无害的乙醇酸。作为可降解生物材料,PGA的力学性能和耐热特性十分突出,是目前已知可降解生物材料中降解性能最好的高分子材料,且具有海水可降解性。面对石油危机和海洋、土壤中的不可降解塑料(微塑料颗粒)的污染,针对PGA材料的研究具有重大意义。但与目前主流的生物基材料聚乳酸(PLA)相比,PGA合成成本高、加工性能差、脆性强,因此,目前PGA主要以纤维形式应用于医疗领域。对PGA改性的意义更多在于将其常规化,通过改性将成本降低应用于日常生活中,以谋求解决塑料污染问题,使其优异的降解性能得到利用。
1 PGA的合成及全球产能分布
1.1 PGA的合成
PGA的合成方法有很多种,最简单直接的是一步或两步缩聚法。用乙交酯(GA)单体直接缩聚合成PGA(图1)[1],但是用这种方法合成的PGA分子量普遍较低(2万以下)。因PGA的熔点与反应温度接近,容易发生热降解;在缩聚反应时会产生水,这也限制了最终产物的分子量。为了提高PGA的分子量,常用开环聚合法来生产PGA(图2)[2],国内外在这方面研究也较多。乙交酯开环聚合先合成低分子量PGA,然后再高温裂解成六元环乙交酯,最后加入引发剂发生聚合反应合成PGA[2]。这种方法制备的PGA比直接缩聚法分子量高很多,可达十万甚至百万级别,但是成本高昂,未规模化应用。
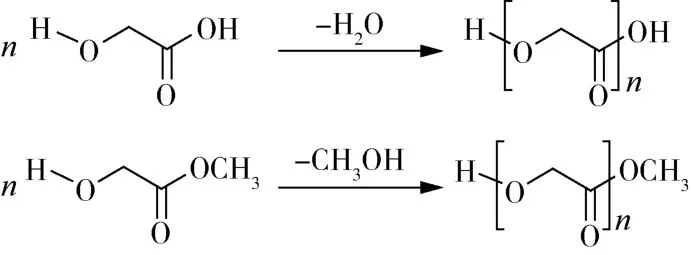
图1 直接缩聚法制备PGA[1]Fig.1 Preparation of PGA by direct polycondensation[1]
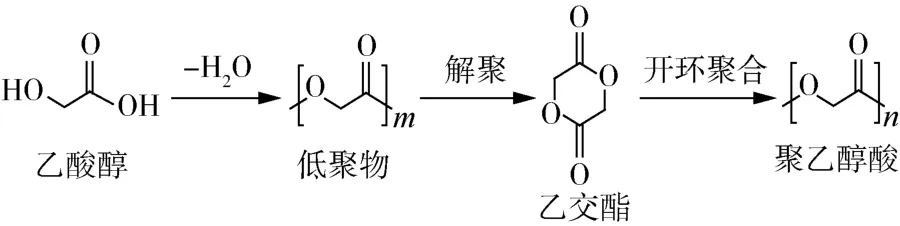
图2 开环聚合法制备PGA[1]Fig.2 Preparation of PGA by ring opening polymerization[1]
1.2 PGA全球产能分布
全球PGA产能尚未达到万吨级规模。PGA规模化生产起始于日本吴羽公司,从1995年开始将PGA批量化生产,也是最早研究出PGA量产技术的公司。到目前为止,日本吴羽也一直领航于全球PGA产业化技术,在2002年和2008年吴羽公司分别在日本投建了产量100 t/a的工厂、在美国杜邦公司投资1亿美元建设4 kt/a的设备,目前已经生产了多种牌号的PGA。国内方面,目前我国现存规划的PGA年产能只有3 kt,近两年国家“限塑令”的出台直接促进了我国石化领域越发注重对PGA材料的研发和应用。以中国石化等央企为首,推动国内PGA工厂建设。据报道,2020年国家能源集团投资10亿元建设产量50 kt/a的PGA产线;同年中国石化也投资了产量200 kt/a的工厂,目前已经在建设当中。此外,上海浦景化工也投资建设了产量10 kt/a的煤基PGA生产工厂。
2 PGA改性
2.1 PGA的物性
PGA难溶于大部分有机溶剂,但溶于在高氟化强极性的溶剂,如六氟异丙醇(HFIP)、氟丙酮基多巴等。其热熔融温度Tm=220~230℃、结晶温度Tc=160~200℃、玻璃化转变温度Tg=35~40℃。由于PGA的两条分子链以正交排列,结晶状态下为全反式构象,因此PGA表现出较高的力学性能以及较高的热变形温度(≈170℃)[3]。但由于其热降解温度为230~250℃,从熔点温度开始PGA熔体就会发生分子内的酯交换降解[4]。同时,较高的结晶温度使得PGA熔体稍微受到冷却即流动困难,加工温度窗口较窄,在实际加工过程中极易因热氧作用导致酯键断裂而降解,且在注塑成型中易因注塑喷嘴冷却而堵塞注射孔。
与其他可生物降解聚合物相比,PGA的立体规整性也使其本身更加坚硬。表1比较了PGA与其他可生物降解性聚合物的热、力学性能[6-18]。由此可见,PGA材料较高力学性能和耐热性能甚至比普通塑料和工程塑料更加突出。将PGA材料与通用工程塑料如高密度聚乙烯(PE-HD)、聚丙烯(PP)、聚酰胺11(PA11)及特种塑料聚醚醚酮(PEEK)相比可以看出(图3),PGA所表现出了突出的耐温性与力学性能,几乎媲美于PEEK,其强度与耐温性同比其他生物降解材料优势明显。但也存在断裂韧度偏低、脆性强的弱点,因此常需要对其进行增韧改性。

图3 不同材料的耐温性及韧性比较[5]Fig.3 Comparison of temperature resistance and toughness of different materials[5]

表1 PGA和其他可降解高分子材料的力学性能比较Tab.1 Comparison of mechanical properties between PGA and other degradable polymer materials
2.2 PGA改性的必要性
如前所述,尽管PGA具有100%可降解性等优势,但是加工性能差、加工温度高、加工温度窗口窄、脆性强、货架期短等不足也导致PGA材料的产业化应用存在障碍,且至今国内方面也未建立起完善的PGA材料加工理论体系及专用加工设备系统。其中,加工过程中材料极易发生热氧降解是最主要的原因,而关于PGA材料的改性机理也并无系统性的报道。所以,尽管PGA材料的机械强度、降解性很突出,但是高成本、低韧性的不足却限制了其推广使用,因此常需要通过改性来实现其对加工环境的适应及性能的改善。
3 PGA熔融改性方法
在对PGA材料进行热熔加工改性时,需要特别注意以下事项:
(1)PGA降解峰值温度(≈250℃)接近Tm(220~230℃),因此要十分谨慎地在不引起热降解的情况下进行加工,尤其是要精确控制熔体加工过程中剪切导致的黏性耗散温升。如可通过添加抗氧稳定剂、增塑剂、扩链剂等,以期提高材料的大分子的缠结网络,改善耐热性、流动性以及水下降解特性[19-20]。
(2)PGA在含水条件下降解速度很快,在250℃以上温度条件下降解更快,因此不适于长时间热熔加工。Sato等[21]通过用抗水解稳定剂(N,N-2,6-二异丙基苯基碳二亚胺)改变端基结构并减少残余乙交酯的量,该方法可将PGA的水解速率减慢了一个数量级。因此,PGA的热熔加工一定要充分干燥。
(3)PGA不溶于大多数有机溶剂。在室温下,PGA只能溶解在HFIP等高氟化溶剂中,在共混改性时一般需要使用相容剂改善其与改性材料的相容性。
PGA的改性方法包括熔融共混、溶液共混以及共聚改性等。其中,PGA熔融混炼是主要的改性手段,是将PGA、改性材料及改性助剂以一定比例混合均匀,通过挤出机或密炼机进行混炼,使改性后的材料同时兼备PGA与改性材料的性能,从而弥补PGA材料韧性的不足。PGA熔融共混改性主要是为了增韧、增强、增塑,下面将从这3个方面讨论PGA的熔融共混改性方法。
3.1 PGA增韧改性
关于PGA增韧改性的研究较少,主要是由于PGA材料本身成本高而尚未规模化民用生产。根据分子结构相似原理,受启发于脂肪族聚酯PLA物理方法增韧改性,我们可以借鉴这些方法来增韧PGA。如图4所示,PGA和PLA的大分子组成几乎相同,只是PLA比PGA多一个甲基。因此,提高PLA的韧性是其应用的必要条件,而PGA也是如此。

图4 PGA及PLA的分子结构Fig.4 Molecular structure of PGA and PLA
聚环氧乙烷(PEO)是也一种可生物降解的聚合物,且生物相容性良好。据报道,PEO是适合PLA的增韧剂[22]。由此可尝试用PEO来增韧PGA。Chang等[23]使用将PGA与PEO共混来提高PGA韧性,将不同含量的PEO与PGA熔融共混,并研究了PGA/PEO共混物相之间的热性能、机械性能和形态结构。结果发现即使PEO含量增加,PEO和PGA之间也不会发生交联,且傅里叶红外光谱(FTIR)分析表明,PEO是通过物理作用实现了对PGA的增韧。DSC数据表明引入PEO会产生稀释效应,使PGA和PEO两相之间的热力学行为相互影响,这说明两种物质的互容性,但PEO过量时则会引起宏观相分离。因此,PGA/PEO共混物韧性的增加可归因于具有适当相容性的PGA/PEO共混物中连续PEO相的形成和演变,故PEO可用于PGA韧性的改善。
PGA本身就是一种优异的可生物降解材料,为了不影响其降解性能,在改性的时候也需要用其他可降解材料用来对其改性,如PBAT、PLA、PBS、PCL、PHA、PVA、PHB、聚甲基乙撑碳酸酯(PPC)以及热塑性淀粉等高分子材料。其中,PVA一种具有良好水溶特性的材料,可使改性后的材料具备更优异的水溶降解性能,而其他的材料大多为聚酯类可降解材料。例如,PBAT是一种可降解生物基弹性体材料,Christopher等[24]通过熔融挤出将PGA与PBAT共混改性,并选用乙烯-丙烯酸甲酯-甲基丙烯酸缩水甘油酯(EMAGMA,缩写为EMAG)作为增容剂。结果发现,共混物的相形态、流变性、热性能、力学性能和阻气性能均得到极大改善。当三种物质PGA、PBAT、EMAG以50/50/20质量比共混时,复合材料表现出较高的拉伸强度(10.5 MPa)和断裂伸长率(145.2%)。此外,Christopher等认为增容剂的引入提高了共混物的整体结晶度,添加质量分数分别为20%和30%的EMAG后,复合材料体系的氧气渗透性和水蒸气渗透性均有所降低。同样地,Niu等[25]也使用EMAG作为增容剂,通过反应共混制备了熔体强度增强的PGA/PBAT共混物。首先将EMAG与PBAT混合,然后与PGA混合形成PGA-g-EMAG-g-PBAT共聚物。这些共聚物选择性地分布在PGA和PBAT之间的界面处,如此有效提高了界面附着力和相容性。当受到拉伸或冲击时,共聚物还充当桥梁,将更多的有效能量从PGA基体转移到PBAT相,从而产生高增韧效果。流变学和动态力学测试结果证实,PGA的熔体强度和玻璃化转变行为因共聚物的形成而得到改善。由此可知,EMAG作为优异的增容剂可显著提升PGA和PBAT材料的相容性,少量EMAG便可改善PGA的力学性能。此外,含活性基团的扩链剂也可用于PGA材料的混炼改性,如Shen等[26]在制备PGA/PBAT共混物时引入ADR作为相容剂与扩链剂,结果发现共混物韧性提高但强度下降。因此,需要调控好ADR的使用量,需要对共混物的界面演变进行深入研究。
3.2 PGA增强改性
虽然PGA的力学性能与其他可生物降解材料相比表现出优异性,但是在某些情况下还是无法满足需求,所以需进一步通过增强改性来大幅提高其力学性能。PGA的增强改性是将PGA与其他材料的表面通过物理或化学吸附形成分子级别上的交联结构,而增强效果与材料的相容性有关。
在对聚合物材料进行增强改性时要考虑两种材料的相容性,一种有效的增容方式是先将要共混的材料中的一种材料与传统相容剂进行接枝,形成的聚合物作为改性要用的相容剂,然后再进行混炼,这样共混材料的界面相容性可以得到明显的改善。贾天飞等[27]研究了PP与PGA共混时的相容性,先将PP与马来酸酐(MAH)接枝形成共聚物(PP-g-MAH)作为共混相容剂,然后再将PP与PGA混炼。因PP-g-MAH中存在酸酐极性基团,可使界面形成交联结构从而改善其相容性,从而得到了相容性较好的共混材料。何敏等[28]也研究了PP、PGA以及相容剂分别在不同配比时的力学性能,发现混合相不均匀会造成材料力学性能下降。两种材料并不会完全相容,需要加入偶联剂加强材料的结合力。Wan等[29]将PGA和PBAT以质量比为20∶80的比例共混制备薄膜,加入质量分数为2%EMAG使PGA/PBAT共混物增容,EMAG通过环氧化物基团与PGA及PBAT的链端基团反应,连接了共混组分之间的界面,EMAG与大分子链的反应可以通过酯交换和环氧化物开环反应进行。经电子束处理后的共混材料具有高应变、高强度和高坚韧的特点。杨海存等[30]用乙烯-丙烯共聚物/凹凸棒土(EPC/ATP)作为相容剂,也制备了PP/PGA共混物,并进一步将PP/PGA共混物颗粒干燥后熔融纺丝得到纤维,然后用玻璃纤维(GF)增强。相容剂质量分数为5%时收缩率处于一个较低的状态,此状态下PP/PGA(质量比为93/7)的共混物表现出最低的收缩率2.1%,而GF改性纤维则大大增强了材料的强度,70%含量时拉伸强度达291.5 MPa。
3.3 PGA增塑改性
为了提高纯PGA材料的热稳定性,扩大其加工温度窗口,提高PGA材料在近熔点附近的塑化能力,在加工过程中可加入一定的增塑剂来防止分子链过热降解,同时引入扩链剂和抗氧剂,扩链剂是通过补充加工过程中因为过热降解而损失的分子链来保持分子链的完整性,抗氧剂是通过与活泼的自由基反应形成稳定的化合物终止连锁反应从而减少分子链降解[31]。
Chen等[32]采用双螺杆挤出机混炼法,将反应性扩链剂4,40-亚甲基双(苯基异氰酸酯)和苯乙烯-丙烯酸多功能环氧低聚物同时加入PGA中。通过引入苯基异氰酸酯和苯乙烯-丙烯酸多官能环氧化物低聚物(CEs)来改变PGA的分子结构。随着扩链剂用量的增加,分子链延长,PGA分子量增大,熔体流动速率越来越慢。剪切流变学数据显示,其弹性模量和复数黏度显著增加,弛豫时间延长,表明分子结构中存在长链分支。改性后的PGA塑化性能有明显改善,熔体拉伸性能也产生了应变硬化现象。因此,CEs不仅对控制PGA的降解有着重要作用,还对改善流变性能也有促进作用。此外,改性后的材料也具有一定的抗水解性能。段雪蕾等[33]也采用抗氧剂和扩链剂协同对PGA进行热塑改性,发现优良的复配体系对PGA的热氧稳定性有很大的提升。乔虎[34]等通过加入扩链剂来改善PGA与PLA共混界面的相容性,在PLA和PGA共混时加入不同比例的扩链剂ADR-4730F,结果表明加入扩链剂后共混材料的力学性能有明显提高,说明加入扩链剂后界面相容性变好。Wang[35]等在制备PETG/PGA共混物时也引入多环氧ADR扩链剂来提高界面相容性、增加熔体黏度并改善流变性能。结果发现PETG/PGA(65/35)的共混物力学性能最佳,拉伸性能随断裂伸长率的增加而增强。扩链剂的加入会导致PGA结晶度降低,不同浓度的ADR会对共混物的性能有不同的影响,因此可以通过调控ADR的加入量得到不同性能的共混物。
综上可知,PGA熔融共混加工一般需要加入特定的相容剂来改善多相材料间的界面相容性,如此增强了相界面会使混炼改性更容易,而塑化性能提升的混炼体系使混炼剪切过程中的黏性耗散程度减弱,局部温升下降也会有效缓解PGA材料的热氧降解。PGA改性所用的相容剂多为环氧类化合物,其中使用最为广泛的相容剂是EMAG和ADR,它们除了具有优异的增容效果还具有增塑效果。
3.4 PGA溶液共混改性
PGA溶液共混主要是将PGA溶液与改性材料的溶液以一定的比例混合均匀,然后通过铺膜、纺丝或絮凝等手段制备出复合体系的产品。如静电纺丝PCLPGA共混纤维可以在相态上将PCL与PGA混合均匀。对PGA而言,静电纺丝是制造具有受控形态、力学性能和降解分布的高纵横比纤维的实用方法[36-39]。
Shayla等[40]通过静电纺丝产生的可混溶的PCLPGA共混纤维的使用,以增加纤维与聚合物-聚合物复合材料的PCL基体的界面结合,制备了PCL/PCLPGA共混复合材料纤维。向PCL基质中添加质量分数为40%的PCL-PGA共混纤维显示出拉伸屈服强度和弹性模量的增大。随着屈服强度和模量的增加,复合材料的断裂伸长率也有所下降。在不使用增容剂的情况下,用PCL基体和PCL-PGA共混纤维增强物获得了韧性更强的长期可生物降解复合材料。
Young等[41]将PGA/PLA共混物(质量比分别为90/10、70/30、50/50、30/70)静电纺丝形成超细纤维,其中PLA通过选择性溶解去除,形成具有三维互联孔的纤维,差示扫描量热(DSC)分析表明,PLA和PGA之间存在相分离的形态结构。
3.5 PGA共聚改性
PGA的共聚改性通常是从分子结构上对PGA材料进行改善,本质上讲,此时共聚合成的材料是一种全新的材料,但由于保留了原来PGA合成时的一些分子单子,因此也就赋予了改性材料部分PGA本身的性能特征。如PLA可以和PGA实现共聚改性,PLA有左旋结构和右旋结构,左旋PLA单体可以和PGA单体共聚合成PLGA;右旋PLA单体可以和PGA单体共聚合成PGLA,两者有不同的结构所以具有不同的性能[42]。Li等[43]利用开环聚合制备了立构规整排列有序的PLGA聚合物,结果发现相比于随机排列的PLGA,简单交替有序排列的PLGA聚合物的总体降解速率较慢且具有更好的热性能。Ajioka等[44]发现了一种可以在溶剂中制备PLGA的方法,但是成本较高无法实现批量应用。PLGA的制备也可以用熔融聚合的方法,此法制得的PLGA分子量一般不高。工业上一般采用PLA单体和PGA单体开环聚合的方法批量生产PLGA[4]。PCL也可以和PGA通过它们的单体共聚合成新的材料,新材料具备两种材料的优势-高强度和可降解性[45]。赵伟君等[46]通过微波熔融聚合法将PGA和纳米晶纤维素(NCC)共聚制成了PGA-NCC,通过改变NCC的加入量制成了不同比例的PGA-NCC,PGA的基本结构没有发生改变,NCC含量越高共聚物的Tg升高,但是结晶度、熔点降低,含量为20%NCC的共聚物强度最优,但此时结晶度下降明显。因此,共聚改性是在分子结构、单体的比例、排列规则程度上实现对PGA材料的改性,从而实现调节聚合重复单元组成以及分子量完成对材料的改性[47]。
综上,我们可以基于不同的方法实现对PGA材料的增韧、增容、增强、增塑等改性,从而实现PGA材料在应用性能及加工特性方面的改进。其中,各种改性方法所期望解决的问题存在一定的差异,如表2所示。同时,在解决实际问题时,可能这些改性策略会复合采用,实现增韧、增强又提高加工性能的目标。

表2 PGA改性方法的优缺点及效果Tab.2 Advantages,disadvantages and effects of PGA modification
4 PGA的水下降解特性
4.1 PGA的水环境降解机理
赋予生物基聚合物材料水溶降解特性有利于彻底解决海洋中的微塑料球污染问题,真正做到全生命周期、全天候条件下可降解,避免大量不可见微塑料在生命体内的沉积。水分子会造成PGA分子链中酯键的断裂,生成含有羧基和含羟基的链段,是缩聚反应的逆反应。PGA与PLA结构相似,二者水解过程也类似。因为PLA多一个甲基导致亲水的酯键发生水解反应比PGA慢,所以PGA的水环境降解性优于PLA,且PGA水解产生的乙交酯还会促进PGA的降解,如图5所示。其中PGA在碱性和中性条件下的水解过程一致,首先发生分子内酯交换反应,羟基端与羰基亲核反应生成六元环作中间体,水解过程中碱与羟基端基的相互作用还具有一定的催化效应。PGA在酸性条件下的降解则是从羟基端质子化开始,然后形成“氢桥”现象,在这一过程中乙醇酸会从低聚乙醇酸中分离出来[48-49]。

图5 PGA在酸性、碱性条件下水解过程[48-49]Fig.5 Hydrolysis of PGA under acidic and alkaline conditions[48-49]
4.2 溶液中PGA的降解
目前,PGA材料作为可降解生物材料中水下降解特性最好的材料之一,也是少数几种海水可降解的材料之一。因此,水下降解特性赋予了PGA材料非常广阔的应用前景,这是海水不可降解材料如PLA、PBAT等无法比拟的绝对优势。
PGA可在磷酸盐溶液中发生降解,检测到结晶度在早期显着增加而后期逐渐降低。这表明PGA的降解首先发生在非晶区域,降解方式为水解降解与裂解诱导结晶,然后进一步结晶区发生降解[50-51]。因此,PGA的水解降解分为两阶段,第一阶段在降解前期并不会发生急剧降解;第二阶段是一段时间后(约7 d),开始急剧降解。发生这种情况的原因可能是一开始是无定形区降解,之后再是结晶区开始降解此时有明显变化。因此,在水解降解过程中,材料的结晶度会发生变化。结晶度先增大后逐渐降低。降解过程中结晶度的变化也必然加速材料本身的力学性能失效。
4.2.1 PGA纤维降解过程中质量损失和形态变化
脂肪族聚酯材料的降解一般是酯骨架在水性条件下的简单水解而发生[52-54]。降解率的高低取决于结晶度、分子量、共聚物组成、形态结构等。You等[50]比较了电纺制备的PGA、PLA和PLGA纳米纤维在磷酸盐缓冲溶液中的降解特性,如图6所示。结果表明,在不存在前期诱导期的情况下,PGA的降解率要明显高于PLGA与PLA,20 d后残留量约40%,作者认为这是由于PGA纤维具更高的比表面积而引起的更快降解。
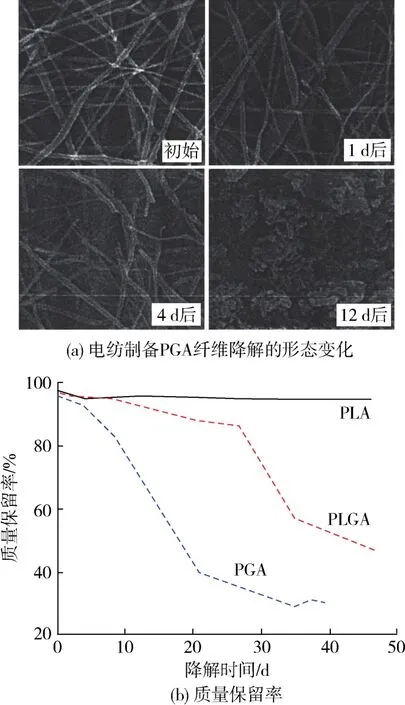
图6 电纺制备PGA纤维降解的形态变化及PGA、PLA和PLGA的质量损失[50]Fig.6 Morphological changes of degradation of PGA fibers prepared by electrospinning and the quality loss of PGA,PLA and PLGA[50]
从电纺PGA纤维的降解形态变化可以看出,即使在短短的1 d后,部分PGA纳米纤维就已经开始分解,并形成局部位置处的纤维断裂,从而使纤维的力学强度丧失。而大量的纤维分解需要4 d的时间,继续浸泡12 d后,PGA纳米纤维则基本全部水解成碎末颗粒。
4.2.2 PGA降解过程中pH值、断裂强度和结晶度的变化
PGA有亲水性是因为其大分子链中含有亲水可以和水发生反应的酯基,其水下降解过程一般是由不稳定的化学键的断裂引发,而后逐渐发展到整个大分子链的断裂,且降解过程中分离出来的乙醇酸会促进PGA降解。
郭正等[55]将PGA纤维及编织线置于37℃、pH=7.4的磷酸盐缓冲溶液中进行生物性水下降解实验的研究,其发现溶液pH值先变小后变大最后逐步恢复,如图7(a)所示。这可能是因为初始PGA初始降解产生的酸性物质多,所以pH值减小,但随着降解过程的继续推进,材料质量损失率逐渐减小,故降解产生的酸性物质变少,所以pH值又逐渐恢复到原来水平。而这一过程所导致的材料力学强度随着降解时间的推移而持续降低,如图7(b)。这表明,尽管PGA的水解过程会呈现出结晶、质量损失快慢及降解产物导致的水环境酸碱性变化,但材料本身的力学性能却几乎是现象衰减。

图7 PGA降解过程中溶液PH的变化和断裂强力的变化[55]Fig.7 Change of pH and breaking strength of PGA solution during degradation[55]
图8表示了降解过程中PGA材料内部结晶度的变化。一般认为,PGA的初始熔融焓为139 J/g,通过DSC测试出降解后材料熔融焓的变化可以初步分析出材料结晶度的变化。结果表明,前2 d内PGA样品的总体结晶度稍有增大,但之后结晶度快速减小。首先,由于材料质量几乎没有减少,所可以认为PGA材料在降解过程中诱导了非晶区域向结晶区域转变,非晶态区域的缠结链降解成碎片,促进了大分子链的有序排列;其次,少部分的PGA材料内部的非晶区域首先发生裂解,导致原先结晶区域及诱导的新结晶区域占残留材料整体的含量比要提高,从而表观上也得到材料的结晶度增大。当非晶区域部分降解至一定程度后,结晶区也逐步发生降解。当然如前所述,这种水下降解过程与材料与水环境的接触比表面积相关,如PGA超细纤维的降解就要比PGA纳米纤维慢得多[56]。

图8 PGA纤维的结晶度随降解时间的变化[50]Fig.8 Change of crystallinity of PGA fiber with degradation time[50]
4.3 海水中PGA的降解
PGA在堆肥环境下的降解速率比较快,只需120 d即可完全降解,在此环境下与纤维素类似,如图9所示。此外,PGA在海水中降解速率也能达到纤维素的降解速率,在海水中28 d后降解可达75.3%[57]。但是,我们有时需要改性调控PGA在水环境下的降解速率。这可从两方面考虑:(1)在乙交酯合成PGA的过程中与亲水链段或者亲水基团进行共聚,增加改性材料的亲水性来改变其降解速率;(2)与本身降解性能较佳的材料进行熔融混炼改性,如PBAT、生物基淀粉、纤维素、PHA等。

图9 PGA在工业堆肥条件下和在海水中的降解率[57]Fig.9 Degradation rate of PGA in industrial compost and seawater[57]
5 PGA的应用
由于PGA的生物降解性,并最终在生物体内降解成H2O和CO2,因此,这种可降解材料中的“贵族”被逐渐地应用于各个领域。若以应用目标划分,PGA材料在油服、医疗等高附加值领域应用较多,如图10所示。PGA可以制成手术缝合线、心血管支架。PGA可调控的降解率也使其可以用于不同的治疗场景,在可控释放速率药物的载体和组织工程中的应用。与其他材料改性后也可制作人工软骨、食管等假体[58]。

图10 PGA的主要应用Fig.10 Main applications of PGA
在石油天然气开采方面,PGA制成的暂堵绳结、压裂球和桥塞在水压裂应用领域具有天然的优势,这是由于PGA可控的降解速率以及优异的力学性能-可承受70 MPa的静水压力,PGA制成的可降解转向压裂暂堵件已经在页岩油气开采方面得到推广。
除了适用于生物医学、石油开采领域的应用外,PGA的高力学性能和隔气性性能使其在涂层、涂料、膜、热管理、传感、包装和结构等工程应用方面非常有前景。
在民用领域,如薄膜包装行业,PGA薄膜的最大优势在于其不会像产生“白色污染”,且比PLA及PBAT薄膜强度高、抗刺穿性强。PGA还可作为调控降解速率的改性剂,调控各种环境下生物基复合材料的“开关”,PGA与PLA共混材料制成的一次性餐具不但符合卫生标准还耐高温并且比PLA制产品对环境更有利,实现对产品性能释放的最大化[59-64]。因此,随着煤基PGA合成技术的发展,未来PGA必将成为和PLA同等体量的材料。Bonutti[65]制作了一种包装,包装薄膜由两层PGA制成,两层之间插入有反应性化学层。当一层降解时,合成层暴露在空气中并导致剩余层的颜色变化,可以用此来判断保质期到期。
在涂料、涂层行业,PGA具有高效的传热能力,这使其可以当做铁磁加热医疗设备的涂层。该系统可以通过改变导管位置来施加交变磁场实现对血管内的局部加热。PGA还可用于制作抗真菌涂层材料。Boehm[66]等通过结合注塑和绘图光刻技术制造了弹性模量为(9.9±0.3)GPa、硬度为(588.2±33.8)MPa、尖端半径为(25±3)μm的PGA微针阵列。伏立康唑与聚(甲基乙烯基醚-共-马来酸酐)通过喷墨打印沉积在其上,这些微针对白色念珠菌表现出良好的抗真菌性。
6 结语
面对石油资源匮乏以及日益严峻的环境问题,传统的一次性塑料制品将会被可降解材料替代,PGA制品的推广符合可持续性发展理念,为打造低碳排放、环境友好型社会提供了道路。海洋及自然中的塑料微球浓度提高也在警示人们不可见“白色污染”的严重性,对全生命周期可降解材料的开发、应用与推广迫在眉睫,而针对PGA基复合材料的改性研究及应用也会不断进步,为解决塑料污染问题提供途径。
鉴于PGA材料较高的结晶度(45~55%)、优异的热稳定性及气体阻隔性以及极其突出的拉伸强度与弹性模量,且可在堆肥、水下及生物环境中完全降解等优异特性,其作为新一代绿色高分子材料可拓展至PLA、淀粉塑料等传统降解材料无法渗透的海水可降解领域。因此,基于PGA材料的改性研究极其必要,对国家推动“双碳”政策具有重要意义,是未来建设低碳社会不可或缺的绿色材料之一。
猜你喜欢
材料与冶金学报(2022年2期)2022-08-10
建材发展导向(2021年15期)2021-11-05
食品安全导刊(2021年21期)2021-08-30
粉末冶金技术(2021年3期)2021-07-28
能源工程(2021年1期)2021-04-13
烟台大学学报(自然科学与工程版)(2021年1期)2021-03-19
丝绸(2020年6期)2020-06-23
模具制造(2019年7期)2019-09-25
江苏农业科学(2015年1期)2015-04-17
科技创新导报(2014年34期)2015-01-13
