
抗犬细小病毒纳米抗体的制备及其中和活性鉴定
2022-09-22 05:17任泽恒皮雪磊夏安然胡昌辉任桂萍
中国畜牧兽医 2022年9期
任泽恒,皮雪磊,夏安然,孙 悦,胡昌辉,任桂萍
(东北农业大学生命科学学院,生物制药实验室,哈尔滨 150030)
犬细小病毒病是由犬细小病毒(Canine parvovirus,CPV)引起的一种急性病毒性传染类疾病[1],其主要特征为犬出血性肠炎和非化脓性心肌炎,临床表现为呕吐、腹泻、发热和排便带血等症状[2],具有高度传染性和致死性。研究表明,不同年龄不同性别犬均能感染,主要以幼犬最为严重,致死率极高[3]。CPV是一种无囊膜单链DNA病毒[4],含有2个开放阅读框(ORF),分别编码结构蛋白(VP1、VP2)和非结构蛋白(NS1、NS2),其中VP2是最主要的结构蛋白[5-6],能够诱导中和抗体的产生。目前,犬细小病毒病以接种疫苗为主要预防方式[7],但疫苗和临床治疗制剂存在许多缺陷,如弱毒疫苗存在毒株变异和毒力返强以及散毒的风险,母源抗体的影响会导致免疫失败[8-10]。单克隆抗体为鼠源抗体,存在免疫排斥反应[11]等问题。因此,亟需研制治疗效果显著又安全可控的药物。
1993年Hamers在骆驼科动物及软骨鱼类体内发现存在一种缺失轻链和重链第一恒定区(CH1)的抗体,被称为重链抗体(HCAbs)[12]。经过基因编辑技术克隆重链抗体的可变区,得到只由1个重链可变区组成的单域抗体(VHH),也被称为纳米抗体(Nb)[13]。与传统抗体相比,Nb具有免疫原性弱、生产成本低、稳定性强等优势,且犬源抗体相比于鼠源抗体,在治疗时不需要担心免疫排斥等副作用。因此为制备犬源抗CPV的Nb,建立犬源抗CPV pBSD-Nb抗体库对犬细小病毒病的治疗十分重要。王天宇等[14]通过建立纳米抗体库成功筛选出抗猪流行性腹泻病毒S蛋白Nb并进行了中和效价鉴定,抗体效价可达1∶256 000。细菌展示是利用基因工程技术将外源基因与细胞间质分泌型信号肽(NlpA)进行融合表达,将外源蛋白表达在细菌内膜上[15]。N1PA信号肽可以介导外源蛋白穿过细胞,并且在穿越细胞膜的过程中,N1PA信号肽会被降解,最终留下6个氨基酸(CDQSSS)将外源蛋白锚定在细菌内膜外表面[16-17]。本研究旨在建立犬源抗CPV pBSD-Nb库,并通过细菌展示结合流式细胞术检测筛选出具有高亲和力的Nb,为抗CPV基因工程抗体药物筛选提供依据,也为研制中和活性全长抗体奠定基础。
1 材料与方法
1.1 材料
大肠杆菌Rosetta(DE3)PlysS感受态细胞、大肠杆菌DH5α感受态细胞均购自北京全式金生物技术有限公司;CPV、CPV高免比格犬脾脏、血清、Fitc标记的VP2蛋白、pBSD展示载体均由东北农业大学生命科学学院生物制药教研室制备并保存。猫肾细胞(F81)购自中国科学院上海细胞库(保藏号:19375.09.3101MAMGNO13)。RNA提取试剂盒购自QIAGEN公司;pMD19-T、pET-27b载体均购自Novagen公司;SfiⅠ、NdeⅠ、XhoⅠ限制性内切酶、PCR试剂、T4 DNA连接酶、DL2000 DNA Marker、IPTG、溶菌酶、抗生素均购自TaKaRa公司;HRP-Sheep Anti-Dog IgG(H+L)购自Abcam公司。
1.2 方法
1.2.1 pBSD-Nb库构建 取CPV高免比格犬的脾脏剪成小块至预冷的研磨管中,每管1 mg,按照动物组织总RNA提取试剂盒说明书提取RNA并反转录合成cDNA。根据GenBank中犬重链可变区(VH)基因的序列,利用Primer Premier 5.0软件设计引物,引物信息见表1。引物均由北京擎科生物技术有限公司合成。以cDNA为模板扩增抗体库VH基因,引入SfiⅠ酶切位点,分别对VH和pBSD载体进行SfiⅠ单酶切。利用T4 DNA连接酶将VH和pBSD载体片段4 ℃过夜连接。将PCR产物电转化大肠杆菌DH5α感受态细胞,涂布于LB固体培养基(含有50 μg/mL氯霉素),在37 ℃条件下培养12 h,构建pBSD-Nb库。

表1 pBSD-Nb库引物
1.2.2 pBSD-Nb库筛选 挑取1.2.1中的单菌落至LB液体培养基中,37 ℃培养至D600nm值约0.4后,加0.25 mmol/L IPTG,37 ℃、120 r/min诱导4 h。诱导结束后,取1 mL培养物12 000 r/min离心1 min,弃上清;加入1 mL PBS洗涤,12 000 r/min离心1 min,弃上清;用350 μL Tris蔗糖溶液(含蔗糖0.26 μmol,Tris 0.04 mol)重悬菌体;加入35 μL新配制的溶菌酶至终浓度1 mg/mL;在涡旋振荡仪上缓慢滴加700 μL预冷的EDTA溶液(1 mmol/L),混匀后冰上静置15 min;加入50 μL预冷的MgCl2溶液(0.5 mol/L),混匀后冰上静置10 min;12 000 r/min离心3 min,弃上清;加入1 mL PBS溶液洗涤原生质体,4 ℃、8 000 r/min离心3 min收集菌体。取90 μL的PBS重悬原生质体。向制备的原生质体中依次加入9 μL 1%牛血清白蛋白(BSA)溶液,1 μL荧光标记的VP2蛋白(FITC-VP2),冰上避光孵育1 h,8 000 r/min离心3 min,离心后收集菌体,用PBS洗涤3次,离心后弃上清,用PBS重悬沉淀。通过流式细胞仪检测阴性对照在488 nm波长激光下激发的荧光强度,收集样品中荧光强度大于阴性对照的部分。提取所筛选细菌的质粒,将质粒进行电转,完成抗体库的构建。继续按照上述方法对抗体库进行2轮、3轮、4轮筛选,直至抗体库阳性率达到80%以上。此时即可挑取固体培养基平板上的单菌落,将所挑取的单菌落分别培养,用流式细胞术逐一检测单菌落,将获得的阳性克隆送北京擎科生物技术有限公司测序。
1.2.3 pET27b-Nb重组质粒的构建及鉴定 根据测序结果,通过Primer Premier 5.0软件设计测序结果正确的Nb基因的上、下游引物,上游引入NdeⅠ酶切位点,下游引入XhoⅠ酶切位点,引物序列及酶切位点序列信息见表2,引物均由北京擎科生物技术有限公司合成。对FCM检测为阳性克隆的质粒中Nb基因片段进行PCR扩增,PCR产物进行1.0%琼脂糖凝胶电泳检测,将大小约380 bp的片段切下进行胶回收。将pET27b载体和Nb基因用NdeⅠ、XhoⅠ双酶切,获得酶切产物用T4 DNA连接酶4 ℃过夜连接。将连接产物转入大肠杆菌DH5α感受态细胞,37 ℃过夜培养,挑取单菌落鉴定正确的质粒送北京擎科生物技术有限公司进行测序。取10 ng测序正确的阳性重组质粒转化大肠杆菌Rosetta(DE3)PlysS感受态细胞。涂布于LB固体培养基上(含50 μg/ mL 卡那霉素),37 ℃培养12 h。

表2 Nb引物信息
1.2.4 Nb基因蛋白的表达与纯化 挑取鉴定正确的大肠杆菌Rosetta(DE3)PlysS菌落接种到LB培养基中(含有50 μg/mL卡那霉素),37 ℃、120 r/min振荡培养4~6 h,当菌液D600 nm值达到0.4时,加入IPTG诱导剂,37 ℃、120 r/min诱导4 h,收集诱导菌,经超声破碎后离心,分别取上清液和沉淀进行SDS-PAGE分析。取1 mL上述菌液扩大培养。4 ℃、4 000 r/min离心30 min收集诱导后菌体,用适量PBS重悬菌体,加入溶菌酶至终浓度为1 mg/mL,冰上静置1 h后超声破碎,4 ℃、12 000 r/min离心30 min,弃上清,利用包涵体洗涤液(尿素浓度为2 mol/L)对包涵体进行洗涤,洗涤2次后,用包涵体变性液(尿素浓度为8 mol/L)重悬包涵体蛋白,将变性蛋白逐滴加入复性液(尿素浓度为2 mol/L)中。将复性后的蛋白置4 ℃ PBS溶液中透析18 h,分装后―80 ℃保存备用。将透析后的蛋白进行SDS-PAGE分析,并用紫外分光光度计检测,计算蛋白浓度。
1.2.5 ELISA法检测Nb对CPV的亲和力 用包被液将Nb分别稀释至100、50、20、10、1、0.1 μg/mL,包被于96孔板,每孔100 μL,置4 ℃过夜。包被过夜后用PBST洗涤5次,每次3 min。使用5%脱脂奶粉37 ℃封闭3 h,洗涤5次后,加入CPV稀释液100 μL,置于37 ℃孵育1 h,洗涤5次后,加入1∶9 000稀释的抗CPV阳性血清作为一抗,37 ℃孵育1 h,洗涤5次,1∶7 500稀释的HRP-Sheep Anti-Dog IgG(H+L)二抗中37 ℃孵育1 h,洗涤5次,加入100 μL TMB底物液避光显色15 min,每孔加入50 μL终止液,酶标仪测定各孔D450 nm值。设置只加底物的PBS组为空白对照(N1)、未加CPV的为阴性对照(N2)、用小鹅瘟病毒(GPV)代替CPV的为条件对照(N3)。样品及对照均设置3个重复。
1.2.6 CPV滴度测定及Nb中和活性检测 将96孔板中的F81细胞培养至汇合度达80%,吸去上清,用DMEM将CPV按101~108梯度稀释后加入F81细胞中,每孔100 μL,每个稀释度设8个平行孔。同时设置只加入100 μL DMEM培养液的正常细胞为对照组,在37 ℃、5% CO2培养箱中培养。每天观察记录细胞病变效应(CPE)。利用Reed-Muench法测定病毒的半数组织培养感染剂量(TCID50)。
将Nb按照21~29的梯度用DMEM进行稀释,分别与100 TCID50CPV混合后37 ℃孵育1 h。将F81细胞接种到96孔细胞培养板上,培养至汇合度达80%,吸去上清,加入Nb-CPV混合液,每孔100 μL,每个稀释度设4个平行孔。同时设置DMEM代替混合液的空白组(C1),只加CPV病毒液代替混合液的病毒组(C2),加抗GPV-Nb代替CPV-Nb的GPV-Nb组(C3)。将细胞置于37 ℃、5% CO2的培养箱中进行培养,每天观察并记录CPE,连续7 d。利用Reed-Muench法对Nb的中和效价进行测定。
1.3 数据统计与分析
用GraphPad Prism 5.0软件进行单因素方差分析,用t检验进行组间差异分析。结果用平均值±标准差表示。P<0.01表示差异极显著。
2 结 果
2.1 pBSD-Nb库的鉴定
由图1A可知,PCR扩增产物大小384 bp,与VH片段预期大小相符。由图1B可知,利用PCR对构建的pBSD-Nb库进行鉴定,获得384 bp的VH片段,证明pBSD-Nb抗体库构建成功。

M,DL2000 DNA Marker;1~8,PCR扩增pBSD-Nb库;9、10,PCR鉴定pBSD-Nb库
2.2 pBSD-Nb库的筛选
流式细胞术检测结果显示,与阴性对照的峰(图2A)相比,含有抗CPV-Nb菌有明显的阳性峰,阳性菌所占的比例为23.8%(图2B),说明部分阳性菌表达的Nb能与VP2蛋白特异性结合。2轮、3轮、4轮筛选结果显示,阳性菌的百分比不断增加(图2C~2E)。第2轮筛选,阳性原生质体比例升至41.9%(图2C);第3轮筛选,阳性率升至61.7%(图2D);第4轮筛选,阳性率达到80.1%(图2E)。流式细胞术检测结果表明,从50株抗体中获得9株阳性峰偏移率较高的抗体,分别命名为Nb1、Nb2、Nb3、Nb4、Nb5、Nb6、Nb7、Nb8和Nb9(图3),将抗体对应的菌株进行测序后,将测序结果于NCBI网站进行Nucleotide BLAST分析,发现测序所得9个Nb序列均与GenBank上已发表的犬抗体VH具有90%的相似性。

A,阴性对照;B~E,第1~4轮筛选

A,阴性对照;B~J,Nb1~Nb9
2.3 Nb基因的克隆
由图4可知,以筛选出的表达9株抗体的质粒为模板,PCR扩增出大小为384 bp的Nb基因特异性目的条带,与预期的大小相符。Nb基因与pET-27b连接后,测序结果显示与Nb原始核苷酸序列一致。
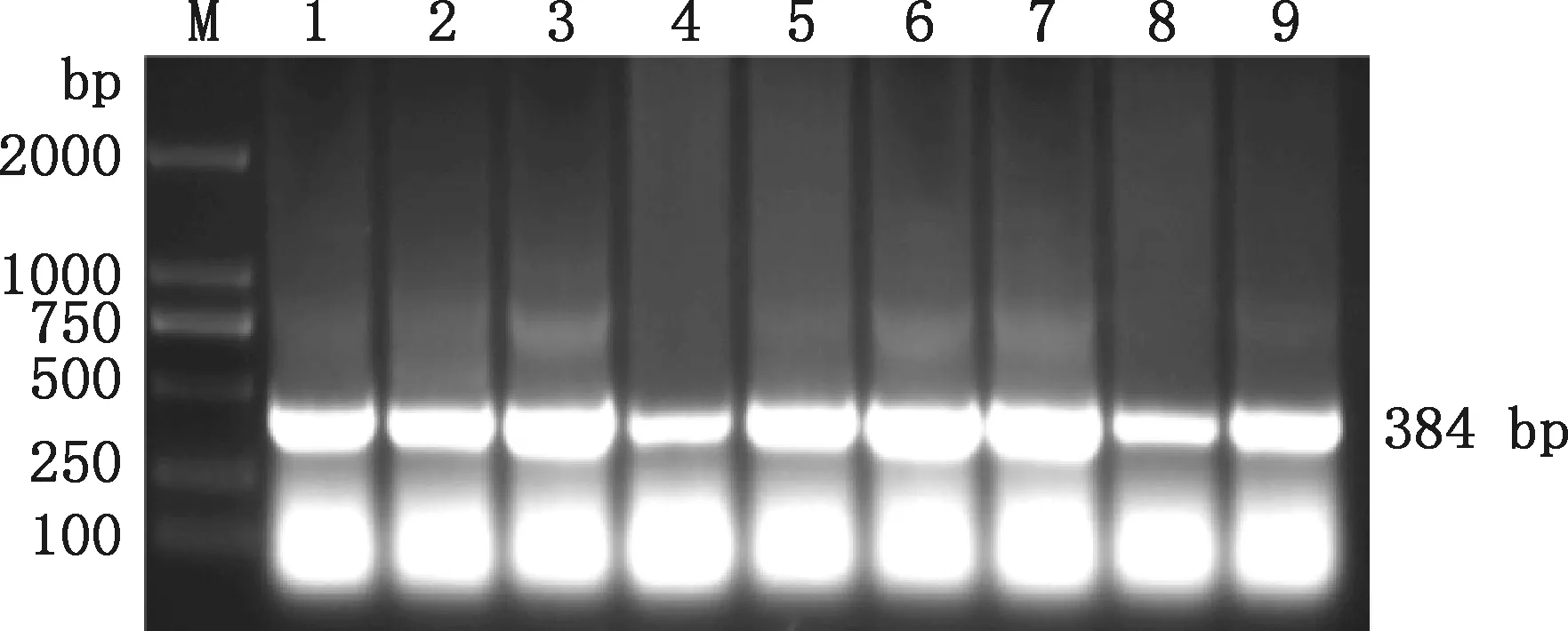
M,DL2000 DNA Marker;1~9,质粒
2.4 Nb蛋白的表达和纯化
对重组质粒pET27b-Nb进行SDS-PAGE检测,结果显示目的蛋白主要以包涵体形式表达,Nb蛋白分子质量约14 ku(图5)。对包涵体变性、复性、纯化及透析,成功获得并表达了目的蛋白(图6),纯化后Nb1~Nb9蛋白的浓度依次为1.65、1.38、1.85、1.60、1.65、1.55、1.46、1.75和1.39 mg/mL。

M,蛋白质分子质量标准;1、5,未诱导阳性菌;2~4、6~11,Nb(Nb1~Nb9)诱导后的沉淀

M,蛋白质分子质量标准;1~9,纯化后Nb蛋白(Nb1~Nb9)
2.5 Nb对CPV的亲和力检测
ELISA检测结果显示,9株抗体的亲和力均极显著高于各对照组 (P<0.01)。随着Nb浓度的增加,反应的光密度值也呈现升高趋势。说明这9个Nb均具有与CPV结合的能力,其中Nb1、Nb2、Nb4、Nb5、N6和Nb9对CPV的亲和力较强(图7)。

与对照组N3相比,**,差异极显著(P<0.01)
2.6 CPV滴度测定和Nb中和活性检测
根据Reed Muench法计算得到CPV的TCID50为10―6.4/0.1 mL。中和试验检测结果表明,9株Nb中有1株具有中和活性,最低有效浓度为0.05 mg/mL(表4)。

表4 Nb中和活性的测定
3 讨 论
CPV于1978年在美国出现[18],并在世界各地犬类中迅速传播,抗CPV抗体的制备一直以来都是研究的热点。本研究建立了pBSD-Nb库,通过细菌展示技术结合流式细胞术对其进行了4轮筛选,成功筛选出9株可以与CPV-VP2结合的Nb。细菌展示技术利用信号肽将外源基因展示在细菌内膜外侧[19],而且锚定外源蛋白的信号肽只有6个氨基酸,对外源蛋白的构象几乎没有影响。细菌周质腔内的氧化性环境,保证了外源蛋白的正确折叠[20],且可以保证蛋白的稳定性,这有利于抗原抗体的结合,保证了抗体筛选的准确性。由于细菌体积比较大,可以通过流式细胞仪进行高通量的分析和筛选,可以快速的富集阳性菌株并筛选出阳性单克隆菌株。
传统方法制备的单克隆抗体,往往在蛋白质的N-端或C-端加入用于鉴定或纯化的标志物,如His标签等。由于其为抗体以外的短肽片段,也许会影响抗体的折叠及功能。本研究直接用无标签的pET-27b载体在大肠杆菌中以包涵体形式表达Nb,经简单处理后,得到纯度和浓度均较高的目的蛋白,减少了去标签纯化的步骤,节约时间,降低纯化损失,并且保证了蛋白能够免受标签的影响,正确折叠[21-23]。
虽然传统单链抗体(scFv)的VH和轻链可变区(VL)片段在连接时的组合是随机的,增加了抗体库的多样性,但也有可能由于VH和VL错配导致scFv活性降低。同时在构建抗体文库的时候scFv经过3次酶切和3次连接,这很有可能使产物在此过程中造成损失。本研究筛选的Nb在结构上比scFv简单很多,它们被单基因编码,不存在由于错配导致活性降低的风险,并且Nb仅经过1次酶切和连接,大大降低了抗体文库在构建过程中抗体的损失,保证了抗体文库的多样性。同时,Nb由于具有较小的分子结构[24],穿透能力强,可以进入致密的组织,使其有可能结合常规抗体不能接近的抗原位点[25]。本试验建立的pBSD-Nb库相比于Pi等[26]建立的pBSD-scFv库,筛选出的Nb的氨基酸序列比scFv的重复率更低,说明pBSD-Nb库相比pBSD-scFv库具有更高的多样性,有更高的几率筛选出具有中和活性的抗体,为日后筛选出更高水平抗CPV的中和抗体奠定基础。
对比ELISA和中和试验结果发现,对病毒具有高亲和力的抗体并不一定具有中和活性。这可能是部分Nb与VP2蛋白非中和抗原表位区段结合导致的。由于无法保证抗体对病毒的亲和力与中和活性的统一,这会使中和活性抗体的筛选效率降低。如果将VP2上中和抗原表位以人工合成的多肽形式进行表达并以其为“诱饵”[27],代替本研究筛选中所使用的VP2蛋白,直接筛选出结合中和表位的高亲和力抗体,即有可能实现抗体对病毒的亲和力与中和活性的统一,CPV中和活性抗体的筛选效率将有可能得到提高。相比于目前一些抗CPV抗体的研究[28],本研究成功获得了1株高亲和力且中和活性更好的抗体,其最低有效浓度为0.05 mg/mL,结果可为抗CPV Nb的制备提供理论依据。
4 结 论
本研究成功构建了抗CPV pBSD-Nb库,筛选出9株对VP2具有高亲和力的Nb,且9株Nb均可特异性结合CPV,6株亲和力较强。其中1株可以阻断CPV对F81细胞的CPE作用,其最低有效浓度为0.05 mg/mL,结果可为构建全长抗体提供参考。
猜你喜欢
中学生物学(2022年8期)2022-10-13
——一道江苏高考题的奥秘解读和拓展
中学生物学(2022年7期)2022-09-07
三农资讯半月报(2020年11期)2020-06-21
江苏农业学报(2019年1期)2019-09-10
智富时代(2016年12期)2016-12-01
智富时代(2016年12期)2016-12-01
戏剧之家(2016年14期)2016-08-02
第二课堂(课外活动版)(2015年3期)2015-10-21
优雅(2015年9期)2015-09-07
意林(2013年19期)2013-05-14
