
施用不同有机物料对砂质土壤玉米成熟期根际细菌群落变化的影响
2022-09-22 11:06:22郝近羽陈源泉代红翠
中国农业大学学报 2022年10期
郝近羽 刘 瑾 陈源泉 代红翠 李 超 徐 洁 隋 鹏*
(1.中国农业大学 农学院,北京 100193;2.山东省农业科学院 作物研究所,济南 250100;3.吴桥县科技局,河北 沧州 061800)
土壤微生物活跃于陆地生态系统中,是影响土壤有机质分解、养分循环和病虫害防治等生态功能的关键因素之一[1],对维持土壤生态系统的稳定性以及改善土壤肥力等具有重要作用[2-3]。细菌是数量最多且种群最丰富的土壤微生物,作为生态系统碳、氮、磷循环中的核心环节,土壤细菌通过分解作用对养分元素进行矿化,构成特殊的无机—有机—生物复合体释放到土壤中,推动着生态系统的物质循环和能量流动[4]。作物根际是“作物—细菌—土壤”相互影响最强烈的区域,这种现象被称为根际效应(Rhizosphere effect)[5]。根际细菌群落结构及多样性受还田措施、土壤类型和作物品种等因素的影响,研究根际土壤细菌群落特征对于探究土壤对不同类型有机物料还田措施的响应具有重要意义。
王伏伟等[6]基于Illumina平台Miseq高通量测序技术对小麦分蘖期砂姜黑土耕层土壤细菌进行高通量测序,发现施用化肥会导致土壤pH显著下降、土壤板结,显著降低了土壤细菌的丰富度,酸杆菌门相对丰度最小;秸秆还田显著提高了土壤细菌的多样性,大幅提高了土壤中放线菌门的相对丰度,这可能与秸秆还田处理中全氮含量较高有一定的关系,同时还大幅降低主要参与芳香化合物分解的鞘氨醇单胞菌的相对丰度。王颖等[7]将不同类型和比例的生物炭添加到黄土高原半干旱区土壤中,发现施用生物炭对土壤理化性质产生显著影响,铵态氮是影响细菌群落结构的关键因子,土壤中铵态氮的含量与放线菌门的丰度呈显著正相关,而与绿弯菌门的丰度呈显著负相关。商丽荣等[8]以呼伦贝尔天然羊草草原退化打草场为研究对象,发现45 t/hm2的蚯蚓粪和菌渣混合施用可提高土壤细菌群落的生物多样性,同时发现有效氮、有效钾、有机质和全钾是细菌群落的主要驱动因素。关于秸秆[6]、生物炭[7]以及畜禽粪便[8]等类型的有机物料还田对土壤微生物群落结构和多样性影响的研究已有初步结论,但由于有机物料自身成分的复杂性以及各区域土壤类型多样性,土壤微生物群落受土壤理化性质影响的程度和机理尚无定论。
Illumina高通量测序技术(High-throughput sequencing)被广泛应用于各种土壤环境的细菌多样性研究领域,能够比较真实地反映出环境细菌的代谢及生态响应情况,具有通量高、准确率高、操作简便和成本低等优点[9-10]。近年来,已有较多文献报道了关于我国宁夏回族自治区银北地区龟裂碱土[11]、内蒙古自治区西辽河平原砂壤土[12]、山东省[13]以及云南省山区和半山区[14]等土壤类型对细菌群落多样性的影响,但对于华北平原砂质瘠薄型土壤方面的研究鲜有报道。本研究利用Miseq PE300高通量测序技术以及PICRUSt功能预测分析的方法,同时结合土壤环境因子,分析不同有机物料还田对玉米成熟期根际土壤细菌群落的结构和功能,旨在探究有机物料还田后土壤理化性质对细菌群落变化的影响,以期为该区域合理施用有机物料还田提供参考。
1 材料与方法
1.1 试验地概况
本试验依托中国农业大学吴桥实验站,该实验站位于河北省沧州市吴桥县(37°41′ N,116°37′ E),年均气温13 ℃,年均降雨量562 mm,年均日照时数2 340 h,无霜期201 d,属于温带季风气候。供试土壤类型为砂土,其中砂粒含量84.09%,粉粒含量9.73%,粘粒含量6.18%。长期定位试验始于2012年,试验开始前土壤有机质含量为1.46 g/kg,全氮含量为0.21 g/kg,有效磷含量为4.86 mg/kg,速效钾含量为55.85 mg/kg,pH 8.71。种植模式为冬小麦-夏玉米一年两熟复种轮作,小麦品种为‘济麦22’,条播,播量225 kg/hm2;玉米品种为‘郑单958’,穴播,种植密度为67 500 株/hm2。
1.2 试验设计
采用微区试验方法,先剥离表土层,用水泥围砌成微区,每个微区的面积为4.0 m×5.1 m=20.4 m2,然后将供试土壤均匀地回填到各微区内,回填深度为50 cm。以秸秆(ST)还田为主对照,单施化肥(CF)为副对照,设置了猪粪(PM)、沼渣(BR)和秸秆生物炭(BC)3种有机物料还田,共5个处理,每个处理3次重复,随机区组排列。所用秸秆来自试验地周围普通大田收获的小麦和玉米秸秆,腐熟的猪粪购于当地饲养场,沼渣为猪粪厌氧发酵而成,购于石家庄市泥香生物科技有限公司,秸秆生物质炭购于河南省三利新能源公司。试验地产生的秸秆全部移除,不进行还田。小麦秸秆在玉米苗期覆盖还田,玉米秸秆、猪粪、沼渣和生物炭在小麦播种前翻耕还田,通过3次翻耕使有机物料、无机肥与耕作层土壤混匀,不会扰动底层土壤。
由表1可知,本试验3种有机物料还田处理和ST均以等氮量投入为原则,周年总还氮量均为555 kg/hm2(有机物料还氮量与无机肥施用量的总和)。3种有机物料还田的周年还碳量以主对照(秸秆投入量为小麦7 500 kg/hm2和玉米9 000 kg/hm2,ST)的含碳量为标准,综合考虑其他物料的碳氮比以及农田施用有机肥的实际情况,生物炭处理的还碳量与主对照相同,猪粪与沼渣处理的还碳量均为主对照的1/2。同时,处理组(PM、BR和BC)和对照组(ST和CF)在小麦季和玉米季播种期配施磷肥150 kg/hm2和钾肥150 kg/hm2。
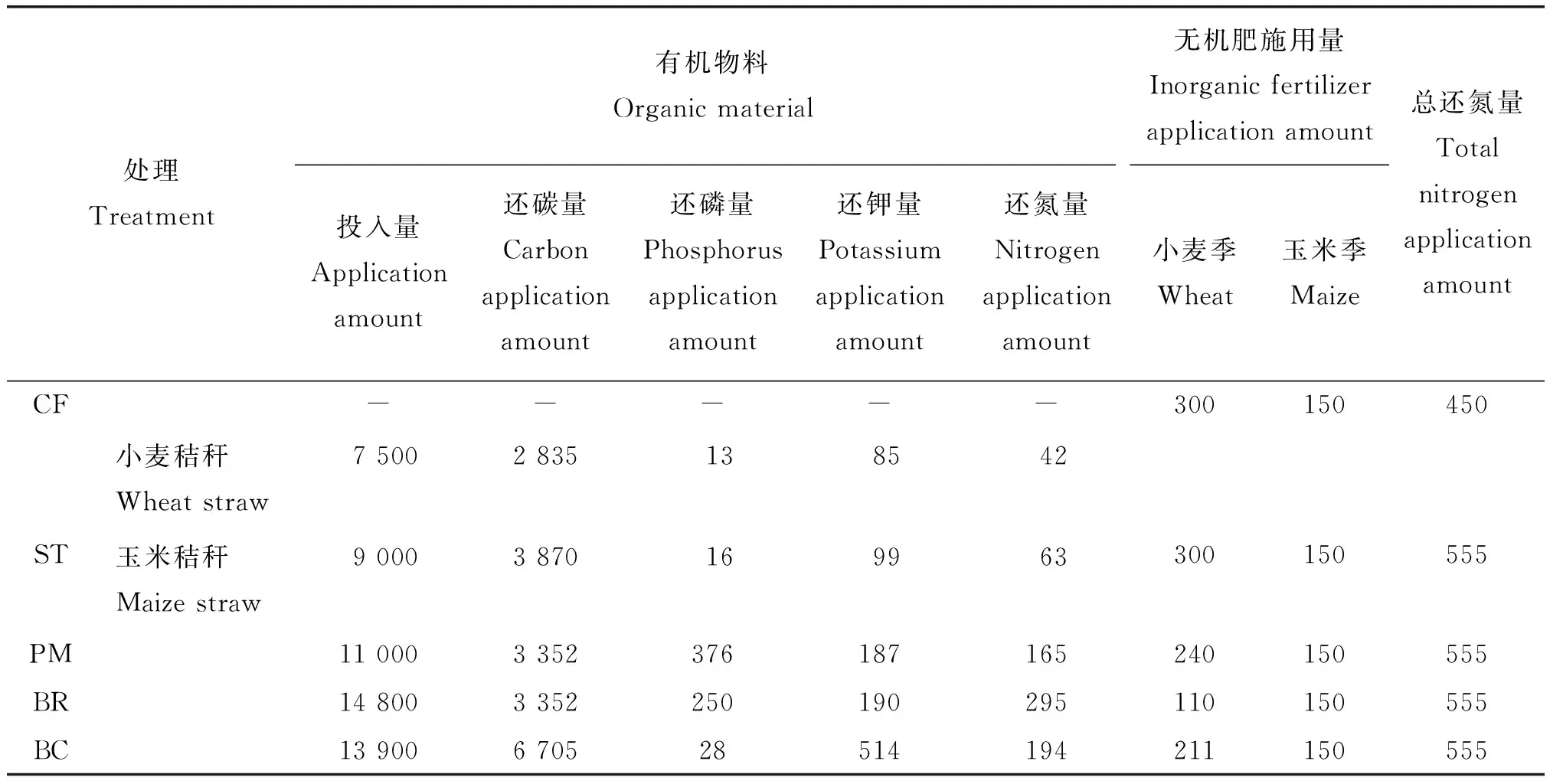
表1 不同有机物料的周年还田量Table 1 Annual application rate of different organic materials amendment kg/hm2
1.3 样品采集
采用五点取样法,于2020年10月(物料还田第8年)收获期时收集玉米根际土壤。采集根际土壤的具体方法:采集时去除玉米根部大块土壤装入塑封袋带内,冰上运输至实验室。然后晃动玉米根部,去除根部松散的表层土壤,使用无菌刷子收集与玉米根系紧密贴合的土壤,即为根际土壤[15]。将同一处理的多点取样土壤样本等量混合均匀后,按试验需求分别保存:用于提取土壤DNA的样品用液氮速冻,置于-80 ℃冰箱保存;用于测定土壤理化性质的样品需风干、研磨。
1.4 土壤理化性质测定
土壤理化性质的测定方法参考《土壤农化分析》[16],土壤全氮(TN)采用凯氏定氮法,有效磷(AP)采用钼锑抗分光光度法,速效钾(AK)采用火焰光度法,有机质(SOM)采用重铬酸钾外加热法,pH采用5∶1水浸-电位法,容重(SBD)采用环刀法,水分含量(SWC)采用烘干法。
1.5 土壤细菌群落多样性测定
用DNA Kit(Omega Bio-Tek,Doraville,GA,USA)试剂盒提取玉米根际土壤DNA,使用1%的琼脂糖凝胶电泳检测DNA的提取质量,核酸蛋白仪(NanoDrop ND-2000)测定DNA 浓度和纯度。选用338F(5′-ACTCCTACGGGAGGCAGCAG-3′)和806R(5′-GGACTACHVGGGTWTCTAAT-3′)对16S rRNA基因V3-V4可变区进行PCR扩增。采用TransGen AP221-02试剂盒,扩增体系为20 μL:5×FastPfu Buffer 4 μL,2.5 mmol/L dNTPs 2 μL,上下游引物(5 μmol/L)各0.8 μL,FastPfu Polymerase 0.4 μL,DNA模板10 ng,ddH2O补至20 μL。于ABI GeneAmp®9700型PCR仪上进行扩增,反应条件如下:95 ℃预变性3 min;95 ℃变性30 s,55 ℃退火30 s,72 ℃延伸30 s,27个循环;72 ℃稳定延伸10 min。每个样本3个重复,将同一样本的PCR产物混合。使用2%琼脂糖凝胶回收PCR产物,利用AxyPrep DNA Gel Extraction Kit(Axygen Biosciences,Union City,CA,USA)进行纯化,并用QuantusTMFluorometer(Promega,USA)进行检测定量。利用Illumina公司的Miseq PE300平台进行测序(上海美吉生物医药科技有限公司)。
1.6 数据分析
原始测序数据使用Fastp(Version 0.20.0)软件[17]对序列进行质控,使用FLASH(Version 1.2.7)软件[18]进行拼接,使用UPARSE(Version 7.1)软件[19]对97%相似水平的OTU(Operational Taxonomic Units)序列进行聚类并剔除嵌合体,使用RDP classifier(Version 2.2)算法[20]对序列进行分类学分析,与Silva 16S rRNA数据库(v138)比对,得到每个OTU对应的分类学信息。
利用Mothur(Version 1.31.2)软件进行α多样性分析,包含丰富度(Chao 1和ACE)和多样性(Shannon)。基于Jaccard距离算法,在考虑共有物种相对丰度的情况下,进行β多样性分析来比较各处理间的物种群落差异。去趋势对应分析(DCA,Detrended Correspondence Analysis)第一排序轴的梯度长度(LGA,Lengths of Gradient)为0.824 8,LGA<3说明土壤细菌群落与环境因子之间适合线性模型,采用Canoco 5进行冗余分析(RDA,Redundancy Analysis),用Duncan’s多重比较方法检验差异显著性,数据处理和作图采用Microsoft Excel 2013、SPSS 20.0和R语言软件。采用PICRUSt2软件进行功能预测分析,与EggNOG(Evolutionary genealogy of genes:Non-supervised Orthologous Groups)和KEGG(Kyoto Encyclopedia of Genes and Genomes)数据库进行比对,获得相关功能和代谢通路的预测信息。
2 结果与分析
2.1 土壤细菌群落多样性
由表2可知,细菌群落覆盖度(Coverage)显示各处理的土壤OTUs测序深度涵盖了土壤中96%以上的细菌,能够比较真实地反映土壤细菌群落的情况。各处理土壤细菌群落丰富度的2个指标(Chao 1和ACE)由高到低均为:PM>BR>ST>BC>CF,说明猪粪、沼渣和生物炭还田后均可提高土壤细菌群落的丰富度,增加了群落内物种的数目。各处理土壤细菌群落的多样性(Shannon)由高到低为:ST>BC>BR>CF>PM,说明不同有机物料还田影响了根际土壤的细菌群落的物种丰富度和均匀度,生物炭和沼渣还田后显著增加了土壤细菌群落多样性,但猪粪还田后显著降低了土壤细菌群落多样性。

表2 不同有机物料还田后土壤细菌高通量测序结果及群落α多样性指数Table 2 Illumina MiSeq sequencing results and α-diversity of soil bacterialcommunity under different organic materials amendment
由图1可知,不同有机物料还田后土壤细菌群落的组间差异大于组内差异(R2=0.937 8)。土壤细菌在OTU水平下的第一主成分(PC1)和第二主成分(PC2)对群落差异性的贡献率分别为 51.22% 和12.90%,累计贡献率为64.12%。对照组(ST和CF)和BC处理均分布在PC1负值区域,且3个处理之间的距离较近,说明相似度较高,而PM和BR处理均分布在PC1正值区域,与上述3个处理之间有较为明显的分离,表明不同有机物料还田后显著改变了土壤细菌群落结构(P<0.001)。

每个点代表一个样品,同处理的样品用同一种颜色表示,同处理的样品连线后形成分组椭圆。Each point in the diagram represents a sample, and the samples of the same treatment are shown in the same color. Group ellipsoid is formed by the connection of the samples of the same treatment.
2.2 土壤细菌群落分布特征
由图2可知,各处理的土壤细菌优势菌门(相对丰度>10%)主要为放线菌门(Actinobacteriota)占18.48%~26.85%、变形菌门(Proteobacteria)占18.79%~26.16%、酸杆菌门(Acidobacteria)占12.12%~17.27%以及绿弯菌门(Chloroflexi)占10.21%~15.73%。此外,BR和PM处理的优势菌门还包含厚壁菌门(Firmicutes)。各处理的优势菌门累计丰度占土壤总细菌群落相对丰度的75%以上,能够在较大程度上代表土壤中主要细菌群落丰度的变化。BR和PM处理的厚壁菌门相对丰度较高,分别为12.11%和26.01%,分别约为ST(6.51%)的2和4倍,但是,BR和PM处理的其余4种优势菌门的相对丰度均有不同程度的降低,尤以PM的最低,其放线菌门和变形菌门的相对丰度比ST和CF分别降低14.51%和31.17%。BC的酸杆菌门相对丰度(17.27%)比ST(12.86%)提高34.29%。
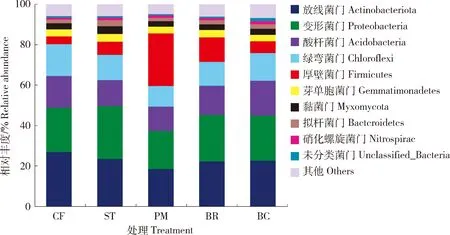
图2 不同有机物料还田后土壤细菌门水平上的物种相对丰度Fig.2 Relative abundance of soil bacterial community based on phylum level under different organic materials amendment
由表3可知,PM的芽孢杆菌属(Bacillus)、梭菌属(Clostridium)和土孢杆菌属(Terrisporobacter)的相对丰度较高,分别为4.51%、6.93%和4.27%,其中,同属于厚壁菌门的梭菌属和土孢杆菌属的相对丰度均显著高于其他4种处理(P<0.05)。BC的盖勒氏菌属(Gaiella)、硝化螺旋菌属(Nitrospira)和类芽孢杆菌属(Paenibacillus)的相对丰度较高,分别为1.56%、1.41%和1.20%,其中,属于放线菌门的盖勒氏菌属的相对丰度比主对照(ST)提高31.67%(P<0.05)。同时,与ST相比,猪粪、沼渣和生物炭还田后显著降低了土壤中类诺卡氏菌属(Nocardioides)和微枝形杆菌属(Microvirga)的相对丰度(P<0.05),此外,还在不同程度上降低了节杆菌属(Arthrobacter)、红杆菌属(Solirubrobacter)、鞘氨醇单胞菌属(Sphingomonas)、芽球菌属(Blastococcus)和链霉菌属(Streptomyces)的相对丰度,但上述丰度下调的菌属在PM、BR和BC之间差异不显著。

表3 不同有机物料还田后土壤主要细菌属的差异性分析Table 3 Differential analysis of soil dominant bacteria genus under different organic materials amendment
2.3 土壤细菌群落与环境因子的关系
由图3可知,在门水平上,环境因子对土壤细菌群落变化的解释率为61.83%,第一排序轴(RDA1)和第二排序轴(RDA2)的解释率分别为51.58%和10.25%,表明环境因子在较大程度上可以解释土壤细菌群落的差异。结果表明,土壤有效磷(AP)对土壤细菌群落分布影响最显著(R2=0.872 1,P<0.05)。
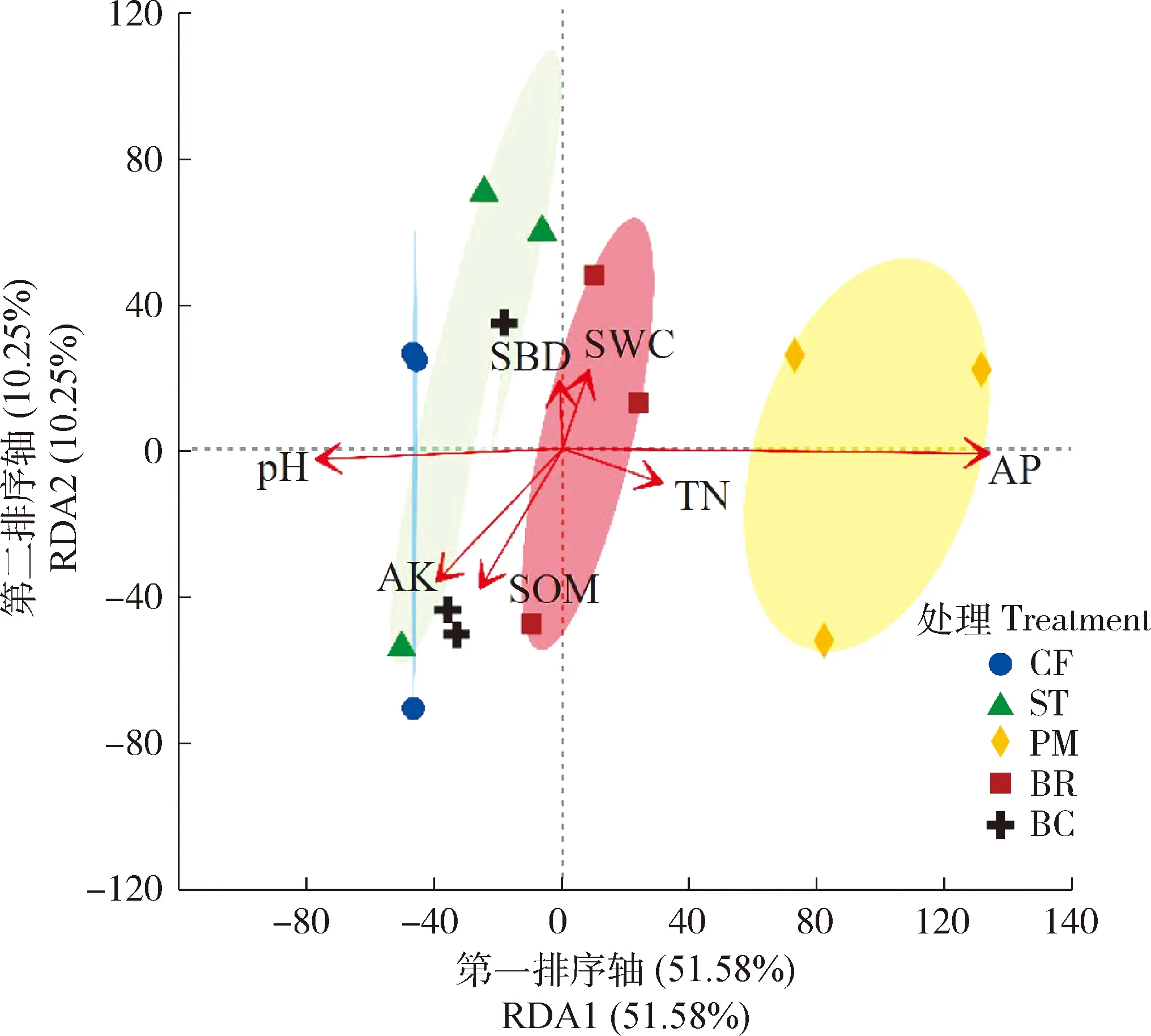
每个点代表一个样品,同处理的样品用同一种颜色表示,同处理的样品连线后形成分组椭圆;图中的每个箭头代表一个环境因子,TN,全氮;AP,有效磷;AK,速效钾;SOM,土壤有机质;pH,土壤pH;SBD,土壤容重;SWC,土壤水分含量。下同。Each point in the diagram represents a sample, and the samples of the same treatment are shown in the same color, group ellipsoid is formed by the connection of the samples of the same treatment. Each arrow in the diagram represents an environmental factor. TN, total nitrogen; AP, available phosphorus; AK, available potassium; SOM, soil organic matter; pH, soil pH; SBD, soil bulk density; SWC, soil moisture content. The same below.
由表4可知,与其他处理相比,猪粪还田后会通过提高土壤有效磷含量进而影响土壤细菌群落结构。此外,与ST相比,BR显著提高土壤全氮含量,还提高土壤水分含量但不显著;BC显著提高土壤pH、有机质、全氮以及水分含量,这可能是造成不同处理间土壤细菌群落差异的重要原因之一。

表4 不同有机物料还田土壤环境因子Table 4 Environmental factors of the soils in different organic materials amendment
由图4可知,土壤有效磷与厚壁菌门的梭菌属(R2=0.753 1,P<0.01)和土孢杆菌属(R2=0.865 2,P<0.001)的相对丰度均呈显著正相关,而与放线菌门的类诺卡氏菌属(R2=-0.532 1,P<0.05)、节杆菌属(R2=-0.534 4,P<0.05)、盖勒氏菌属(R2=-0.707 1,P<0.01)、芽球菌属(R2=-0.760 7,P<0.001)和红杆菌属(R2=-0.771 4,P<0.001)以及变形菌门的鞘氨醇单胞菌属(R2=-0.575 0,P<0.05)、属于酸杆菌门的苔藓杆菌属(Bryobacter)(R2=-0.555 9,P<0.05)的相对丰度均呈显著负相关。土壤pH与放线菌门的盖勒氏菌属(R2=0.700 0,P<0.01)的相对丰度呈显著正相关,土壤容重与放线菌门的类诺卡氏菌属(R2=0.632 7,P<0.05)的相对丰度呈显著正相关,而土壤全氮(R2=-0.608 2,P<0.05)、有机质(R2=-0.589 3,P<0.05)和水分含量(R2=-0.592 9,P<0.05)均与类诺卡氏菌属的相对丰度呈显著负相关。此外,各环境因子还与一些未分类菌门之间存在正、负显著相关性,环境因子对土壤放线菌相对丰度的影响最显著,其次显著改变了厚壁菌、变形菌和酸杆菌等细菌的相对丰度。

*表示P<0.05,**表示P<0.01,***表示P<0.001;越偏红色表示R2越高,越偏绿色表示R2越低。Significant levels are: * P<0.05, ** P<0.01, *** P<0.001. Deeper red indicates higher R2, while deeper green indicates lower R2.
2.4 土壤细菌群落PICRUSt功能预测
通过与EggNOG数据库比对,可以解析出对应的直系同源簇(COG,Cluster of orthologous group)的功能信息和丰度,5种处理共获得4 421个COG编号信息。由表5可知,各处理间土壤细菌群落的主要功能蛋白(酶)的种类基本相同,基因序列注释到催化有机物进行氧化还原反应的脱氢酶/还原酶(COG1028)的丰度最高,其次为注释到一些参与细胞合成/分解及代谢的组氨酸激酶(COG0642)、主要促进剂(COG2814)、NAD依赖性差向异构酶/脱水酶(COG0451)、RNA聚合酶(COG1595)和α/β水解酶(COG0596)等。
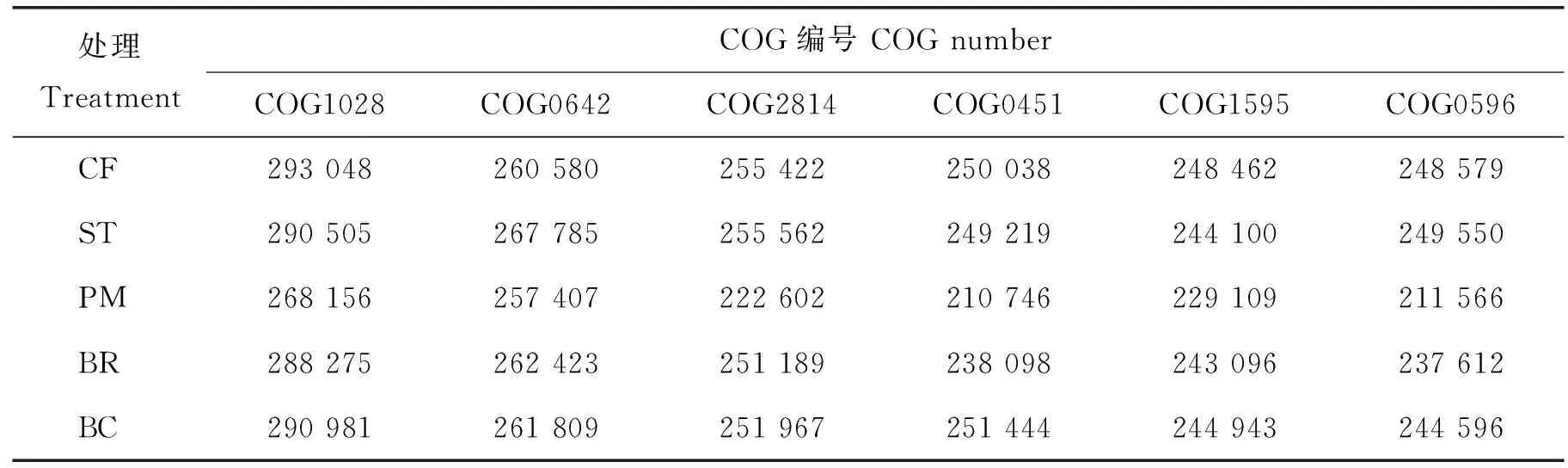
表5 不同有机物料还田后土壤细菌群落主要COG丰度Table 5 The main COG abundance of soil bacterial community underdifferent organic materials amendment 个
由表6可知,5种处理共得到401条代谢通路,按照KEGG代谢通路第一等级将功能分为六大类。基因序列注释到新陈代谢通路的相对丰度最高(77.82%~78.57%),其次为遗传信息加工(6.77%~7.02%)、环境信息加工(5.14%~5.45%)、细胞过程(4.34%~4.50%)和人类疾病(3.34%~3.51%),还有少数基因序列注释到生物体系统(1.81%~1.86%)。由于新陈代谢通路为各处理的主要代谢通路类型,进一步分析其在第三等级下代谢通路的相对丰度。与ST相比,PM、BR和BC的土壤菌群在嘧啶代谢(ko00240)、丙氨酸/天冬氨酸和谷氨酸代谢(ko00250)和肽聚糖生物合成(ko00550)通路均具有优势,增幅分别为2.25%~6.74%、1.33%~4.00% 和3.13%~7.81%;但均降低了不同环境中的微生物代谢(ko01120)和脂肪酸代谢(ko01212)通路的相对丰度,降幅分别为0.79%~2.16%和1.08%~5.38%,见表6。此外,猪粪还田处理的土壤菌群还在丙酮酸代谢(ko00620)、卟啉和叶绿素代谢(ko00860)通路均具有优势,增幅分别为1.85% 和5.56%;但降低了甘氨酸、丝氨酸和苏氨酸代谢(ko00260)、丙酸代谢(ko00640)和柠檬酸循环(ko00020)通路的相对丰度,降幅分别为1.03%、1.28% 和2.70%。

表6 KEGG代谢通路(第三等级)相对丰度的差异性分析Table 6 Differential analysis of KEGG metabolic pathway (level 3) relative abundance
3 讨 论
3.1 不同有机物料还田对土壤细菌群落组成的影响
本研究中,不同有机物料还田后玉米根际土壤细菌多样性和群落结构均发生了显著的变化,猪粪还田后虽然显著增加土壤细菌群落所含物种的种类,但是土壤细菌群落的多样性显著降低,可能是因为猪粪还田后土壤细菌群落分布的均一性较差,不同物种之间所含的个体数相差较大。这与Liu等[21]的研究结论一致,在水稻-油菜轮作种植区施用不同还氮量的猪粪(还田氮量分别为90、180和270 kg/hm2),发现猪粪还田后使水稻季土壤细菌群落多样性下降,且猪粪的还氮量越高,水稻季土壤细菌群落多样性下降的幅度越大。与猪粪还田后的结果相反,生物炭还田后土壤细菌群落所含物种的丰富度较低,但土壤细菌群落分布的均一性较好,还田后可以显著提高土壤细菌群落多样性。而沼渣还田后能够同时提高物种的丰富度和均匀度,进而显著提高土壤细菌群落的多样性。进一步分析发现,各处理的土壤细菌优势菌门的种类相同,均为放线菌门、变形菌门、酸杆菌门和绿弯菌门,因此,土壤细菌群落的多样性变化主要与上述优势菌门的相对丰度密切相关,其中,放线菌门和变形菌门的细菌在所有种类中所占的比例最大。已有研究表明,放线菌门和变形菌门广泛分布于有机质含量高、土壤疏松并且水分条件好的作物根际土壤周围[22-23]。本研究中,生物炭还田后土壤酸杆菌门的相对丰度显著高于其他处理,比秸秆处理增加34.29%,可能是由于生物炭还田后刺激了酸杆菌门在土壤中降解顽固的有机物质的相关代谢活动[24],为酸杆菌门提供了生长机会。土壤中酸杆菌门与变形菌门的数量相当,但由于其难以培养,目前对酸杆菌门在自然环境中的功能还存在争议[25-27]。一些研究表明酸性土壤环境有利于酸杆菌门的代谢活动,因为生物炭自身呈碱性,施入土壤中不利于酸杆菌门细菌的生长[28-29]。但有一些酸杆菌门的基因序列在中性和碱性的环境中被检测出来后[30],进一步研究发现不同施用量或类型的生物炭还田后可以在一定程度上增加酸杆菌门的相对丰度[31-35],尤其是在作物的根际土壤中,酸杆菌门表现出更强的适应能力[36],同时王光华等[24]研究发现酸杆菌门可能参与了土壤的单碳化合物的代谢过程。本研究中,生物炭(BC)和沼渣(BR)处理的绿弯菌门相对丰度分别比养分含量最低的化肥处理(CF)降低13.22%和24.86%,说明有机物料在腐解过程中向土壤释放的养分会在一定程度上抑制绿弯菌门的活性。绿弯菌门也是一类难以培养的微生物,不产氧的光合作用(Anoxygenic photosynthesis)是绿弯菌门固定大气中CO2的最主要方式,这种特殊的光合特性使绿弯菌门以CO2为碳源产生能量时,会在有机质含量较低的土壤中具有竞争优势[37-39]。本研究中,PM和BR的土壤细菌优势菌门还包含厚壁菌门,且PM土壤中优势菌门相对丰度是以厚壁菌门为首,PM可显著增加厚壁菌门的梭菌属和土孢杆菌属的相对丰度。已有研究发现猪盲肠中有众多分解粗纤维的厚壁菌门细菌,尤其是健康仔猪肠道中梭菌属和土孢杆菌属的相对丰度更高,因此,厚壁菌门普遍存在于猪的鲜粪中[40-41]。鲜猪粪在施入农田前还需要堆肥,厚壁菌门是此过程中驱动反硝化作用的关键微生物,能够利用氮源促进猪粪发酵和腐熟[42],猪粪还田会增加土壤中梭菌属和土孢杆菌属等厚壁菌门细菌的相对丰度,为作物提供养分并培肥土壤[43]。
3.2 不同有机物料还田后土壤细菌群落与环境因子的关系
本研究连续8年有机物料还田在很大程度上改变了土壤理化性质,进而影响了土壤细菌群落的形成过程。畜禽粪便自身的磷含量普遍偏高,猪粪还田后可为土壤提供丰富的有机磷[44],并大幅提升土壤厚壁菌的相对丰度[45]。同时,还有研究发现根际土壤厚壁菌门的部分菌属能够将难溶性磷酸盐转化为作物可吸收利用的形态,从而增加土壤中可溶性磷的含量[46],说明土壤环境因子与特定的土壤菌群之间是相互作用的。本研究中,土壤有效磷是影响土壤细菌群落结构变化的主效环境因子,它与梭菌属和土孢杆菌属的相对丰度均呈显著正相关,说明猪粪还田后通过提高土壤有效磷的含量,进而调控并增加土壤厚壁菌门的梭菌属和土孢杆菌属的相对丰度。这与Yang等[47]的研究结论一致,高含氮量的猪粪还田后导致土壤中磷的积累多于其他养分,有效磷含量既是影响细菌群落变化的决定性因素,也是导致高含氮量的猪粪还田后细菌群落多样性下降的重要因素。但是,Huang等[48]研究发现土壤细菌和真菌对于有效磷含量的升高并不敏感,土壤碳循环及化学性质(如pH)的改变可能是磷含量影响细菌和真菌群落结构变化的间接机制。目前关于有效磷含量对土壤微生物影响的研究偏少,本研究中土壤有效磷影响土壤细菌群落结构变化的关键机制也有待进一步的试验验证。
放线菌门广泛分布于土壤环境中,受各环境因子的影响较大,放线菌在中性偏碱的土壤中生长较好[49],增加土壤水分有利于放线菌的生长,土壤有机质与放线菌的相互促进作用明显[50],但李金融等[51]发现放线菌门菌群与有机质含量呈负相关但不显著,上述差异可能与土壤环境条件有较大关系。本研究中,土壤pH与盖勒氏菌属的相对丰度呈显著正相关、土壤有机质与类诺卡氏菌属的相对丰度呈显著负相关。秸秆生物炭是含碳量丰富的多孔性物质,在自然条件下通常呈碱性,还田后会提高土壤pH和有机质的含量[52],说明生物炭还田后提高土壤pH引起盖勒氏菌属丰度显著升高,增加土壤有机质含量使得类诺卡氏菌属丰度显著降低。此外,土壤水分含量也与类诺卡氏菌属的相对丰度呈显著负相关,沼渣还田后改变了土壤水分含量,进而也调控了类诺卡氏菌属丰度。综上,由于放线菌门在属水平下的养分偏好,盖勒氏菌属和类诺卡氏菌属难以培养、营养需求特殊,对土壤养分的调整机理有待进一步研究。
3.3 不同有机物料还田对土壤细菌群落功能的影响
本研究结果表明,各处理的COG功能组成相似,从整体上看,土壤细菌群落细胞中相对丰度最高的是脱氢酶/还原酶,它是糖类、有机酸和氨基酸等物质进行氧化还原反应的重要催化剂,大多数土壤细菌通过此途径获得能量[53]。其次是组氨酸激酶,可与下游靶蛋白一起构成细胞双组分信号传导系统,对土壤细菌的趋化性和密度感应等多种功能有重要作用[54]。然后是参与碳水化合物代谢的主要促进剂、RNA聚合酶和NAD依赖性差向异构酶等功能蛋白协同调控土壤细菌群落细胞的合成、分解及代谢等活动[53]。进一步分析土壤细菌群落与相关代谢的关系,各处理菌群均以新陈代谢的相关通路为主,活跃的代谢活动保证了土壤细菌的存活。与对照组(ST和CF)相比,猪粪(PM)、沼渣(BR)和生物炭(BC)还田处理不同程度的上调了土壤细菌群落的新陈代谢(如嘧啶代谢、丙氨酸/天冬氨酸和谷氨酸代谢、肽聚糖生物合成等),其中,PM对土壤细菌群落新陈代谢通路的影响较为显著。有机物料还田后为土壤带来了较为充足的碳水化合物、含氮有机物、维生素和糖类等物质,为土壤细菌群落的生长提供了新的营养和能量,进而显著影响了细菌群落结构的多样性。但鉴于PICRUSt功能预测分析的局限性,目前只能初步预测出细菌群落的相关功能,后续还需要借助宏基因组测序技术进一步解释影响上述代谢功能的具体原因。
4 结 论
针对华北平原砂质土壤连续8年有机物料还田后玉米成熟期根际土壤研究发现,沼渣和生物炭处理均显著增加了土壤细菌群落多样性,放线菌门和变形菌门是沼渣和生物炭处理的优势细菌;但猪粪处理显著降低土壤细菌群落多样性,厚壁菌门为其优势细菌。土壤有效磷、pH、速效钾和有机质是影响土壤细菌群落结构发生变化的主要因素,其中土壤有效磷对细菌群落分布的影响最显著,与厚壁菌门的梭菌属和土孢杆菌属的相对丰度均呈显著正相关。此外,猪粪、沼渣和生物炭处理均比秸秆处理上调了土壤细菌群落新陈代谢通路中的嘧啶代谢、丙氨酸/天冬氨酸和谷氨酸代谢以及肽聚糖生物合成代谢通路的相对丰度。综上,合理施用沼渣还田可作为改良华北平原砂质瘠薄型农田的首选方式。
猜你喜欢
河南医学研究(2022年19期)2022-10-19 00:44:18
国际呼吸杂志(2019年22期)2019-12-09 09:20:36
生态学报(2019年11期)2019-07-08 06:18:58
纤维复合材料(2018年4期)2018-04-28 08:45:34
石油化工建设(2016年4期)2016-02-27 15:03:16
大型铸锻件(2015年1期)2016-01-12 06:33:06
上海金属(2014年4期)2014-12-15 10:40:46
应用海洋学学报(2014年4期)2014-11-22 07:43:54
天然产物研究与开发(2014年6期)2014-04-27 14:15:56
食品工业科技(2014年23期)2014-03-11 18:19:08
