
氧化联合湿法吸收净化NOx与SO2
2022-09-21 09:57刘恒恒高凤雨易红宏李典泽倪书权唐晓龙
中南大学学报(自然科学版) 2022年8期
刘恒恒,高凤雨,2,易红宏,2,李典泽,倪书权,唐晓龙,2
(1.北京科技大学能源与环境工程学院,北京,100083;2.工业典型污染物资源化处理北京市重点实验室,北京,100083)
NOx和SO2是造成国内外酸雨、光化学烟雾、温室效应等环境问题的主要因素[1]。目前,应用较为广泛的NOx净化工艺为选择性催化还原(SCR)、选择性非催化还原(SNCR)等,SO2净化工艺为密相干塔烟气脱硫法、石灰石−石膏法、氨法等。SO2和NOx分别净化的工艺费用较高、占地面积大、运行系统繁杂,且湿法脱硫后的烟气含有大量水蒸气对SCR 催化剂具有毒害作用[2]。因此,结构紧凑、投资与运行费用低的一体化脱硫脱硝技术研究具有重要意义。
湿法脱硫技术(FGD)是已大规模商业化应用的脱硫方法[3],湿法脱硝技术的前提是将NO 氧化为易溶于水的高价态NOx,因此,NO 的氧化技术是该技术进一步发展的关键,而湿法同时脱硫脱硝技术是在同一吸收系统中同时实现烟气中硫氧化物(SOx)和氮氧化物(NOx)脱除的技术,吸收段脱除效率高[4]。近年来,Mn 基催化剂由于其优良的催化氧化性能而被广泛研究,但在含有SO2的实际烟气条件下,通常有催化剂活性中心硫酸盐化以及气体竞争吸附等问题,导致催化剂活性大幅下降,从而难以实现真正的工业化应用[5]。Ce,Co,Fe和Ni等元素的掺杂可以进一步提升催化剂的抗性、稳定性以及氧化性能[1,6−7],于国峰等[8]发现Ce 的加入可以有效地抑制催化剂活性组分的硫酸化,同时还能降低硫酸盐在催化剂表面的稳定性,从而可以提高催化剂的抗硫性。赵世超等[9]研究发现,金属掺杂改性对Mn/AC 催化剂的抗硫性能有较大提升,而且改性后的催化剂比表面积增加。单纯利用O3直接对NO进行氧化,虽然具有一定的氧化效果,但是运行成本较高。丝梦等[10]发现O3与NO 物质的量比n(O3)/n(NO)<1 时,催化剂的加入显著提升了氧化效率,当反应温度为150 ℃时,联合催化氧化NO 的效率相对于臭氧单独氧化NO的效率提升了约30%。利用催化剂+O3联合氧化技术对NO 催化氧化具有较大的研究意义和应用价值,但目前这方面的报道较少。
因此,本文作者以SO2和NOx为研究的脱除对象,对烟气在除尘后低温条件下的硫硝净化体系进行研究,采用催化氧化联合O3氧化湿法吸收脱除SO2和NOx。为了兼顾环境及经济性的要求,并为了进一步提高催化剂的氧化以及抗硫中毒性能,利用元素掺杂的方式对催化剂进行改性,并且通过叠加O3氧化,在减少氧化剂用量的同时降低SO2对脱硝效果的影响。
1 实验
1.1 实验装置
采用钢瓶气模拟烟气[2],其中,NO体积分数为400×10−6,SO2体积分数为100×10−6,O2体积分数为5%,平衡气为N2,反应空速为31 000 h−1。实验系统示意图如图1所示。实验系统包括配气系统、氧化系统、吸收系统和分析系统4 个部分,其中,氧化系统分为催化氧化和O3直接氧化2 个部分。采用1 L 的锥形玻璃容器作为氧化反应器,装有0.05 mol/L Ca(OH)2乳浊液的1 L鼓泡反应器作为液相吸收装置。采用KM9106便携式综合烟气分析仪(Kane)检测进出口气体成分,Model106-M 型臭氧分析仪检测O3体积分数。

图1 实验系统示意图Fig.1 Schematic diagram of experimental system
1.2 催化剂制备
以(NH4)2CO3为沉淀剂,采用共沉淀法制备Mn0.4Ce0.6Ox和Mn0.4Ce0.5MyOx催化剂(M 代 表Ni,Co,Cu,Fe,Sn,Mg 及空白),沉淀剂与金属离子物质的量比为1 :1。金属阳离子前驱体Mn(CH3COO)2·4H2O(纯度99.0%),Ce(NO3)3·6H2O(纯度99.0%),Cu(NO3)2·3H2O(纯度99.0%),Co(NO3)2·6H2O(纯度98.5%),Ni(NO3)2·6H2O(纯度98.0%),Fe(NO3)3·9H2O(纯 度98.5%),Mg(NO3)2·6H2O(纯度99.0%)和SnCl4·5H2O(纯度99.0%)均为分析纯,采购自国药集团。
催化剂制备过程中,首先,将前驱体溶解于200 mL 去离子水,超声波处理10~20 min,将0.5 mol/L 的(NH4)2CO3溶液缓慢滴加至前驱体溶液中,并机械搅拌(老化)1 h;对搅拌后溶液进行过滤,并收集滤纸上的固体过滤物,用去离子水反复洗涤3~4次后放置于烘箱中,在120 ℃干燥12 h后;然后,将样品在350~550 ℃条件下煅烧6 h;最后,将制备好的催化剂粉碎并过筛至0.425~0.850 mm,用于催化剂性能测试。
1.3 测量与分析方法
在催化氧化过程中,以NO 转化率η(NO)为氧化性能评价指标,其计算方法如下:

式中:φin(NO)和φout(NO)分别为进口、出口NO 体积分数,10−6。
湿法吸收以及组合工艺的过程中,以脱除率作为脱硫脱硝的性能评价指标。NOx脱除率η(NOx)和SO2脱除率η(SO2)分别为:

式中:φin(NOx)和φout(NOx)分别为进口、出口NOx体积分数,10−6;φin(SO2)和φout(SO2)分别为进口、出口SO2体积分数,10−6。
2 结果与讨论
2.1 不同锰基复合氧化物催化氧化性能比较
不同催化剂氧化效果对比如图2所示。由图2可知,在250 ℃时,7 种MnCeMOx(M 代表Co,Ni,Fe,Sn,Cu,Mg及空白)催化剂对NO的催化氧化活性性能从大到小依次为Mn0.4Ce0.5Ni0.1Ox,Mn0.4Ce0.5Co0.1Ox,Mn0.4Ce0.5Fe0.1Ox,Mn0.4Ce0.5Cu0.1Ox,Mn0.4Ce0.5Sn0.1Ox,Mn0.4Ce0.5Mg0.1Ox,Mn0.4Ce0.6Ox;在150~200 ℃时,7种MnCeMOx催化剂对NO的催化氧化活性性能从大到小依次为Mn0.4Ce0.5Ni0.1Ox,Mn0.4Ce0.5Co0.1Ox,Mn0.4Ce0.5Cu0.1Ox,Mn0.4Ce0.5Sn0.1Ox,Mn0.4Ce0.5Fe0.1Ox,Mn0.4Ce0.5Mg0.1Ox,Mn0.4Ce0.6Ox。可见,掺杂Ni和Co氧化物的Mn-Ce基催化剂活性较好,在250 ℃时,NO 转化率约50%,与Mn0.4Ce0.5Ox催化剂相比,NO 转化率提升了接近40%。Ni 元素的掺杂增强了反应过程中活性氧的传递效率进一步促进锰和铈活性组分间的协同催化作用,从而加强NO 催化氧化能力[11];而Co 元素的掺杂使催化剂的晶化度降低,有利于质子的嵌入和脱嵌,提供了更多的氧空位,促使催化剂表面氧化还原反应的发生,从而促进了低温催化氧化NO 反应的进行[12]。同时推测,Ni 和Co 元素掺杂会增加催化剂表面强氧化性的Mn4+和Ce4+浓度,进一步促进催化剂对NO 的催化氧化效果[13−14]。这与LI等[7]研究的结果一致。

图2 不同催化剂氧化效果对比Fig.2 Comparison of oxidation effects of different catalysts
高凤雨[15]对Mn0.4Ce0.5Co0.1Ox,Mn0.4Ce0.5Ni0.1Ox和Mn0.4Ce0.6Ox的XRD 谱进行了研究发现,Mn0.4Ce0.6Ox样品在2θ为28.6°,33.0°,47.7°和56.5°处出现4个尖峰,在76.9°,79.1°和88.2°处出现了3 个弱峰,3 个样品均出现了宽反射峰,这可能是由于CeO2萤石结构的存在,同时,Mn 与(Mn+Ce)物质的量比不同,使得XRD 谱的峰位不同[16],在Mn和Ce的混合氧化物中,Ce4+和Mn3+由于结构相似,Mn3+也可能会将CeO2萤石结构中的Ce4+代替。同时,由于Mnn+的离子半径小于Ce4+的离子半径,因此,Mnn+替代Ce4+可能导致单元收缩,从而使CeO2的衍射峰移动到更高的角度[17]。Mn0.4Ce0.5Co0.1Ox和Mn0.4Ce0.5Ni0.1Ox催化剂的衍射峰强度比Mn0.4Ce0.6Ox的衍射峰强度更弱更宽,说明Ni和Co的掺杂使得混合氧化物颗粒变小、结晶度降低,上述这些特性可能有利于提升催化剂的氧化性能。
对Mn0.4Ce0.6Ox,Mn0.4Ce0.5Co0.1Ox和Mn0.4Ce0.5Ni0.1Ox催化剂的物理特征进行BET测试,结果如表1所示。从表1可见:Mn0.4Ce0.5Co0.1Ox和Mn0.4Ce0.5Ni0.1Ox催化剂具有较高的比表面积和孔容,Mn0.4Ce0.5Co0.1Ox,Mn0.4Ce0.5Ni0.1Ox和Mn0.4Ce0.6Ox的比表面积分别为37.36,57.65和31.53 m2/g,孔径分别为7.91,6.84和9.62 nm。研究表明,更高的比表面积有利于反应物的吸附,可以促进催化剂的催化性能[17]。
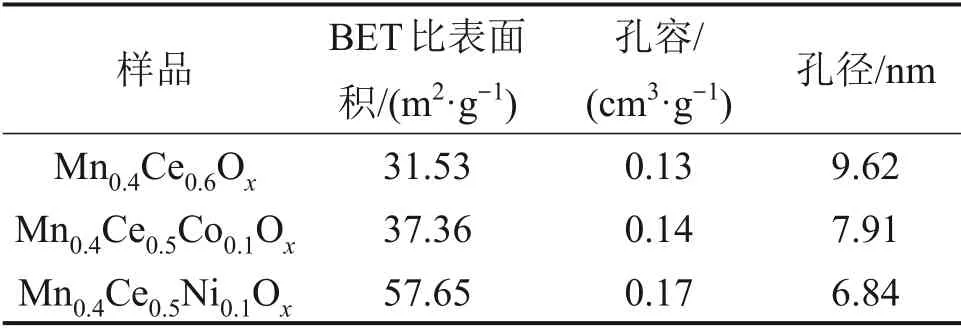
表1 样品的BET比表面积和孔表征Table 1 BET specific surface area and porecharacterization of samples
2.2 焙烧温度对Mn-Ce-Co(Ni)Ox催化氧化NO 性能的影响
不同焙烧温度的Mn0.4Ce0.5Co0.1Ox和Mn0.4Ce0.5Ni0.1Ox催化剂催化氧化效果如图3所示。从图3可见:在反应温度为150~250 ℃,焙烧温度为450 ℃时,催化剂催化氧化效果最佳;在反应温度为250 ℃时,450 ℃下焙烧的Mn0.4Ce0.5Co0.1Ox和Mn0.4Ce0.5Ni0.1Ox催化剂NO 转化率可达到75%左右;而当焙烧温度高于450 ℃时,NO 的转化率却不足50%,表明焙烧温度高于450 ℃时会导致催化剂催化氧化性能下降。因此,焙烧温度对催化剂催化氧化NO 的效果具有较大的影响,2 种催化剂在500 ℃和550 ℃焙烧时,催化剂的催化氧化性能出现整体下降的现象,并且550 ℃焙烧的催化剂催化氧化性能远远低于450 ℃焙烧的催化剂催化氧化性能。因此,推测高温可能会使催化剂烧结,即会使活性组分的晶粒变大,比表面积缩小,导致催化剂活性降低。众多研究结果同样表明催化剂存在热失活现象[18−20],同时较高的温度不利于锰氧化物在催化剂表面的分散[21−22],而焙烧温度过低,催化剂中阴离子可能存在不完全分解的状态,从而导致催化剂焙烧不完全,另外,高温焙烧可能也会导致催化剂表面的MnO2转化为Mn2O3,降低具有高氧化活性的Mn4+含量[23−24]。因此,焙烧温度过高或过低均不利于催化剂催化氧化NO。
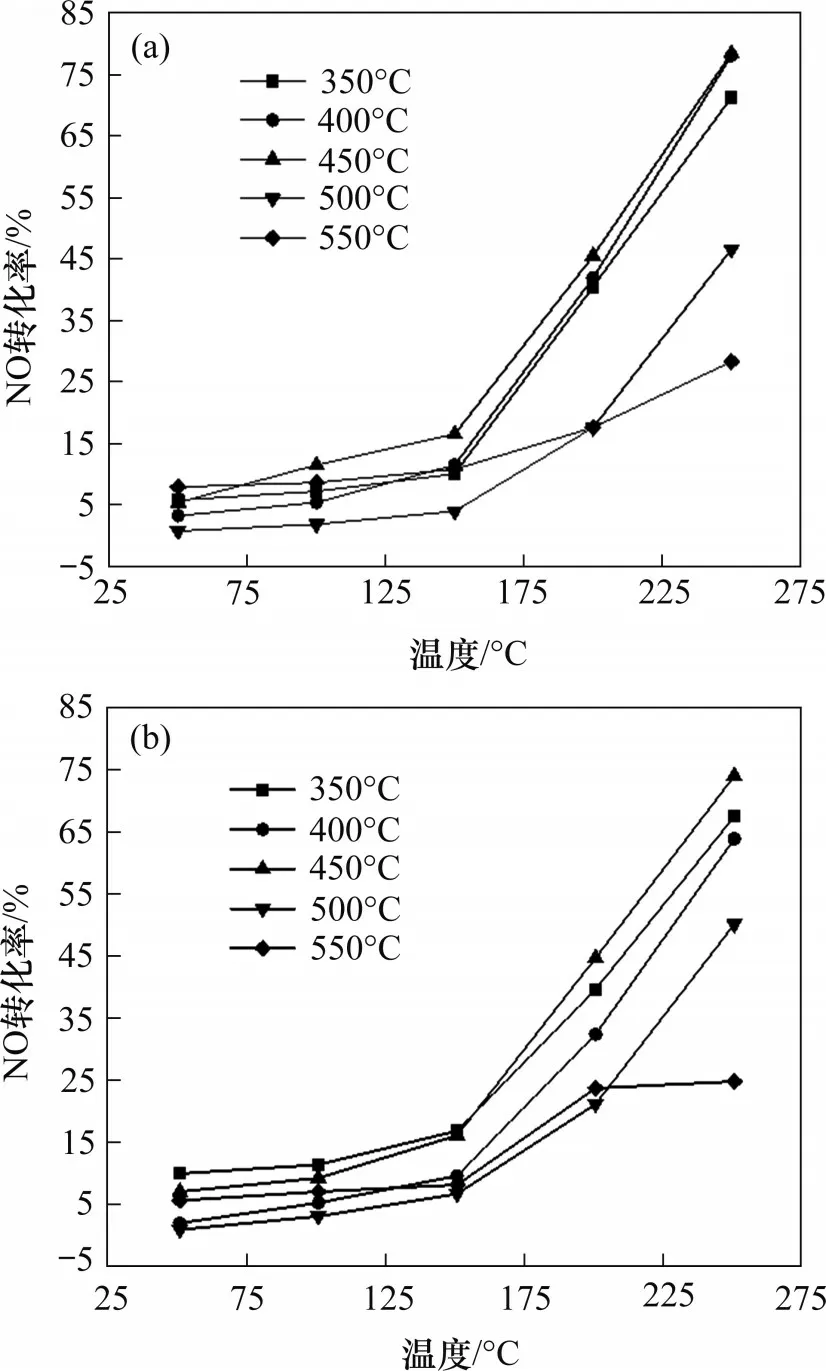
图3 焙烧温度对Mn0.4Ce0.5Co0.1Ox和Mn0.4Ce0.5Ni0.1Ox催化氧化NO性能的影响Fig.3 Effect of calcination temperature on catalytic oxidation NO performance of Mn0.4Ce0.5Co0.1Ox and Mn0.4Ce0.5Ni0.1Ox
2.3 活性组分配比对Mn-Ce-Co(Ni)Ox 催化氧化NO性能的影响
活性组分配比对Mn-Ce-Co(Ni)Ox催化剂(焙烧温度450 ℃)催化氧化NO 性能的影响如图4所示。由图4(a)可知,当反应温度小于200 ℃时,Mn-Ce-CoOx催化剂的NO 转化率从大到小的顺序为Mn0.4Ce0.5Co0.2Ox,Mn0.4Ce0.5Co0.1Ox,Mn0.4Ce0.5Co0.05Ox;当反应温度大于200 ℃时,Mn0.4Ce0.5Co0.2Ox和Mn0.4Ce0.5Co0.1Ox的NO转化率相近,Mn0.4Ce0.5Co0.05Ox的NO 转化率略低。这说明当Co 配比≤0.2 时,在50~250 ℃,Mn-Ce-CoOx催化剂的NO催化氧化性能随Co配比的增加而提高,其中,反应温度为200 ℃时,Mn0.4Ce0.5Co0.2Ox催化剂的NO转化率为43%,反应温度为250 ℃时,其NO转化率达到76%。
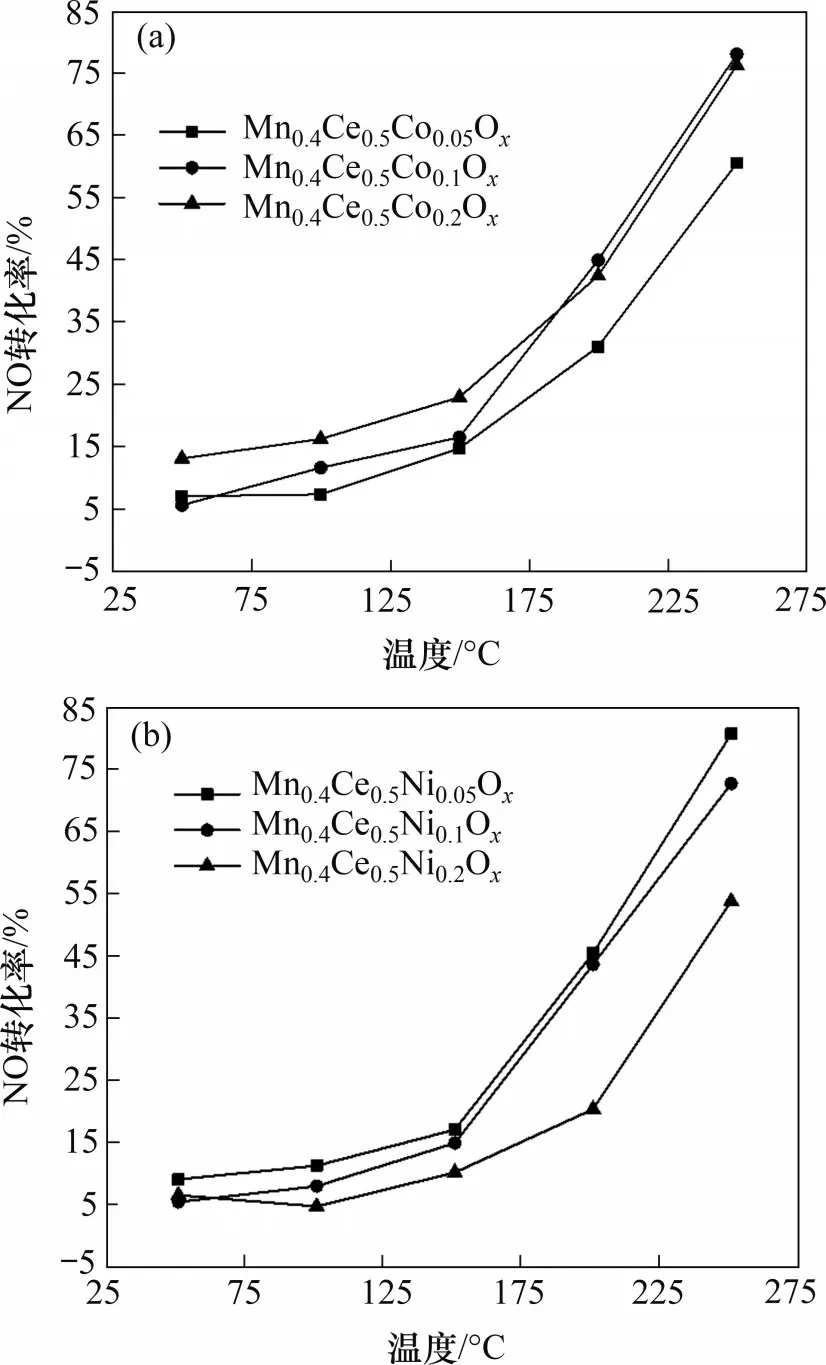
图4 活性组分配比对Mn-Ce-Co(Ni)Ox催化剂(焙烧温度450℃)催化氧化NO性能的影响Fig.4 Effect of active component distribution ratio on the catalytic performance of Mn-Ce-Co(Ni)Ox catalyst(calcination temperature of 450 ℃)for NO oxidation
由图4(b)可知,对于Mn-Ce-NiOx催化剂,3种活性组分配比所制备的催化剂催化氧化效果随反应温度升高而逐渐增强,NO转化率从大到小的顺序为Mn0.4Ce0.5Ni0.05Ox,Mn0.4Ce0.5Ni0.1Ox,Mn0.4Ce0.5Ni0.2Ox。与Mn-Ce-CoOx催化剂不同的是,Mn-Ce-NiOx催化剂的NO催化氧化效果随Ni组分配比增加而下降。当反应温度为200 ℃时,Mn0.4Ce0.5Ni0.05Ox催化剂的NO 转化率超过了45%,反应温度为250 ℃时,其NO 转化率可达82%。这可能是因为Mn 在催化反应中起主活性位作用,而Ce起固定氧的作用[25−27]。Mn0.4Ce0.5Ni0.05Ox催化剂中,Ce与Ni的物质的量比增加,导致氧空位增多[28],有利于提高NO 转化率;而Mn0.4Ce0.5Co0.1Ox催化剂中,Co 元素本身具有很好的氧化活性,适当增大Co与Mn+Ce的物质的量比,有助于催化剂中各组分的协同催化作用[29]。这说明,元素掺杂改性可以提升Mn基催化剂的主要活性组分Mn4+和催化剂表面吸附氧的含量[30],从而进一步促进催化剂的活性。虽然Co 元素的掺杂量增加了1 倍,增加了Co 的成本,但其催化活性并没有明显的提升,因此,在反应温度为150~250 ℃时,Ni 和Co 最优的掺杂比例分别为0.05和0.10。
2.4 SO2对Mn-Ce-Co(Ni)Ox催化性能的影响
Mn-Ce-Co(Ni)Ox催化剂(焙烧温度450 ℃)的抗硫性能如图5所示。由图5可知,在250 ℃和175 ℃的反应条件下,未通SO2时,Mn-Ce-Co(Ni)Ox催化剂NO转化率分别可以保持在80%和40%左右。当通入SO2的体积分数为100×10−6时,SO2在短时间内对催化剂的NO催化氧化性能造成了非常大的影响,50~130 min 内,NO 转化率持续下降,于130 min左右达到最低点。此时,在250 ℃和175 ℃下,Mn0.4Ce0.5Co0.1Ox催化剂NO转化率分别降至约21%和13%,Mn0.4Ce0.5Ni0.05Ox催化剂NO转化率分别降至25%和14%左右;停止通入SO2时,催化剂NO转化率出现略微的回升。这可能是由于SO2与NO存在竞争吸附关系,停止通入SO2,竞争吸附停止,因此催化剂活性出现了轻微的回升,同时SO2的存在会使催化剂活性中心硫酸盐化[31−33],最终在停止通入SO2后,导致催化剂活性未完全恢复,但催化剂还可以保持一定的催化氧化活性。
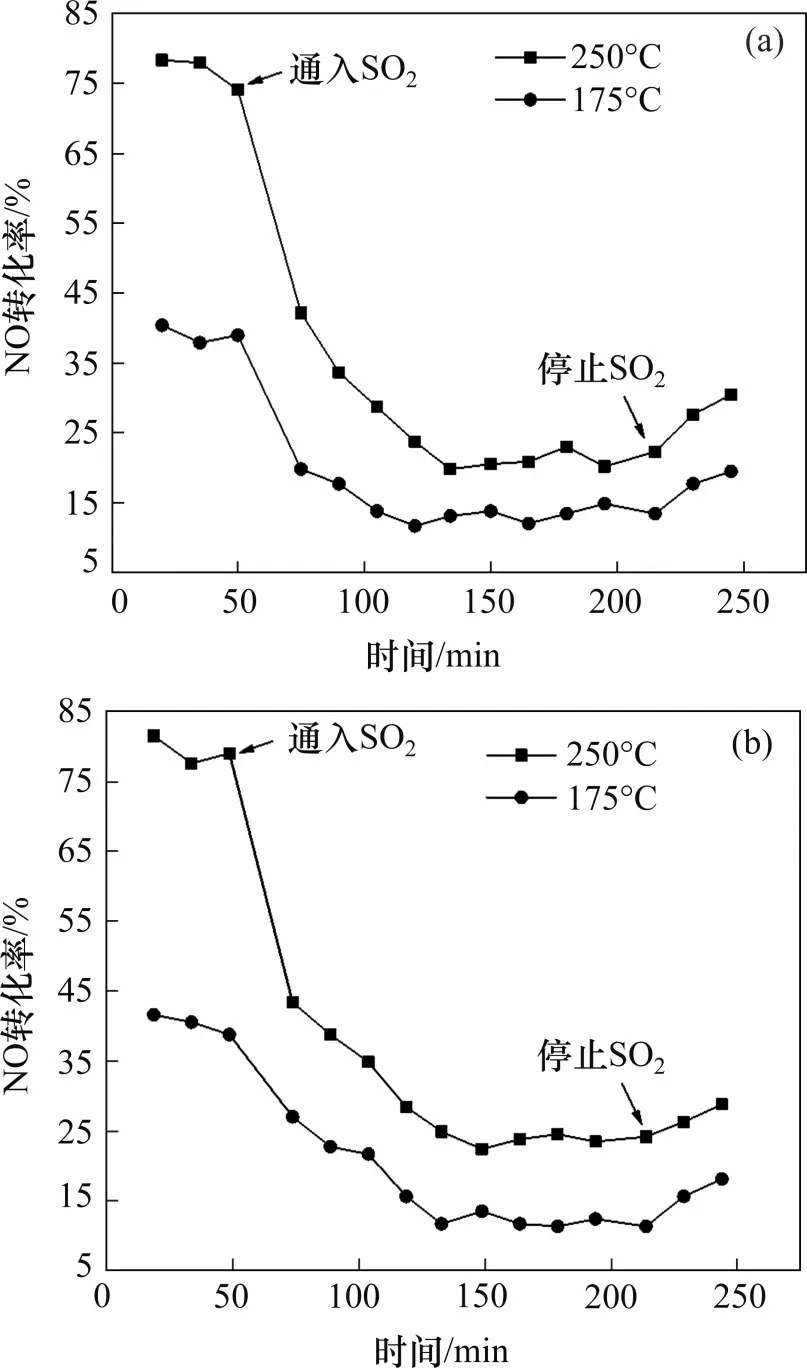
图5 Mn-Ce-Co(Ni)Ox催化剂(焙烧温度450 ℃)抗硫性能Fig.5 Sulfur resistance test of Mn-Ce-Co(Ni)Ox catalyst(calcination temperature of 450 ℃)
2.5 催化氧化+O3氧化联合氧化对脱硝的影响
由于SO2的存在对催化剂具有严重的毒害作用,致使NO催化氧化效果快速降低,从而导致单独催化氧化联合湿法吸收无法实现较高的脱硝转化率。而O3氧化结合湿法吸收时,SO2的存在反而在一定程度上可以促进碱液对NOx的吸收效果[2],但是由于O3氧化运行成本较高,因此,在本实验中,将催化氧化与O3氧化联合,以追求更高的NOx脱除率,同时降低O3用量,减少SO2对最终NOx脱除的影响。根据上述优化结果,采用450 ℃焙烧的Mn0.4Ce0.5Ni0.05Ox催化剂进行催化氧化+O3氧化联合氧化对脱硝的影响的实验。催化氧化+O3氧化与单独的O3氧化效果对比如图6所示。可见,与单独O3氧化相比,联合氧化的NO 转化率更高,NO 转化率达到60%(湿法吸收最佳NO 氧化度)时,O3的用量可减少25%。
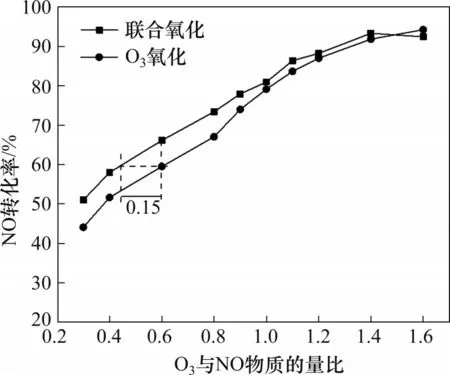
图6 250 ℃时催化氧化+O3氧化与O3氧化效果对比Fig.6 Comparison of effects of catalytic oxidation+O3 oxidation and O3 oxidation at 250 ℃
催化氧化+O3氧化与O3氧化联合吸收的脱硝效果对比如图7所示。从图7可知:在低O3用量(O3与NO 物质的量比小于0.6)段,联合氧化结合湿法吸收的脱硝效率明显要高于单独O3氧化结合湿法吸收的脱硝效率,在O3与NO 物质的量比为0.5 左右时,脱硝效果约提高13%左右,当O3与NO 物质的量比大于0.6,2种方式的脱硝效率较为相近,说明氧化和吸收之间有一定耦合关系。因此,联合氧化方式可行,并且较适用于低O3用量(O3与NO 物质的量比小于0.6)与吸收结合的工艺流程。当O3与NO 物质的量比>0.7 时,单独O3氧化结合鼓泡吸收的方式,就可达到80%的脱硝效果。值得注意的是,研究发现该组合方法中,脱硝效果不仅不会受到硫的负面影响,甚至SO2可以促进吸收液对NOx的吸收效果[2],同时脱硫效果接近100%。综上所述,催化氧化联合O3氧化的工艺可以提升NO的氧化性能,减少SO2对催化氧化系统的影响,并且减少了O3的用量,进一步缩减了成本,减少了温室气体的产生,拥有较多工业生产优势。

图7 催化氧化+O3氧化与O3氧化联合吸收的脱硝效果对比Fig.7 Comparison of denitration effect of combined absorption of catalytic oxidation+O3 oxidation and O3 oxidation
3 结论
1)优化制备了Mn0.4Ce0.5Ni0.05Ox和Mn0.4Ce0.5Co0.1Ox催化剂,2 种催化剂均具有较好的NO 催化氧化效果,这是由于低比例Ni 元素增强了反应过程中活性氧传递效率,高比例Co 元素提升了催化剂固定氧的能力,而且Ni和Co的掺杂提升了催化剂的比表面积,同时增加了催化剂表面高氧化活性的Mn4+和Ce4+含量。
2)Mn-Ce 基催化剂的最佳焙烧温度为450 ℃。焙烧温度过高或过低都会导致催化剂催化氧化性能下降,较高的温度不利于锰氧化物在催化剂表面的分散度、造成催化剂烧结、致使催化剂表面高氧化活性Mn4+减少;焙烧温度过低致使催化剂氧化不完全,影响催化剂结晶度。
3)Mn0.4Ce0.5Co0.1Ox和Mn0.4Ce0.5Ni0.05Ox催化剂在SO2存在时,由于SO2与NO 发生竞争吸附以及催化剂活性中心的硫酸盐化,导致活性下降,停止通入SO2后,可恢复一定的催化氧化活性。
4)与单独的O3氧化相比,联合氧化的NO 转化率更高,NO 转化率达到60%(湿法吸收最佳NO氧化度)时,O3的用量可减少25%。
猜你喜欢
分子催化(2022年1期)2022-11-02
农业工程学报(2022年6期)2022-06-27
无机盐工业(2022年3期)2022-03-11
能源工程(2021年6期)2022-01-06
建材发展导向(2021年16期)2021-10-12
发电技术(2020年3期)2020-06-29
智富时代(2018年3期)2018-06-11
智富时代(2018年3期)2018-06-11
Coco薇(2016年2期)2016-03-22
中学化学(2015年2期)2015-06-05
