
鲈形目TLR1和TLR9基因的选择压力分析
2022-09-17 12:37:50吉红九贾超峰陈淑吟张志勇徐士霞
南京师大学报(自然科学版) 2022年3期
刘 兴,高 波,吉红九,贾超峰,祝 斐,孟 乾,陈淑吟,张志勇,徐士霞
(1.南京师范大学生命科学学院,江苏省生物多样性重点实验室,江苏 南京 210023) (2.江苏省海洋水产研究所,江苏省海水鱼类遗传育种重点实验室,江苏 南通 226007)
鲈形目(Perciformes)是鱼类中物种丰富度最高的一个目,包括25个亚目、160科、1 539属、10 033种[1]. 鲈形目物种分布广泛,大多生活在海洋,仅有少数(如鲈科、丽鲷科等)生活在淡水水域[1]. 鲈形目多个物种是中国重要的海水养殖鱼类,具有重要食用价值和经济价值. 据报道高密度养殖模式会引起鲈形目物种病害频繁发生,尤其是受到多种细菌、病毒等病原微生物威胁[2]. 鱼类病害的爆发会导致其产量和品质的下降,从而造成较为严重的经济损失. 因此,研究鲈形目免疫基因的进化不仅有助于理解宿主和病原体间的作用机制,也为物种遗传育种及疾病防治提供重要的参考依据.
先天性免疫在抵抗外界病原体入侵方面起着重要作用,是动物的主要防御机制,代表着抵御病原体入侵的第一道防线,在维持生物体生长、发育和生存过程中发挥最重要作用[3-4]. 鱼类属于变温动物,其淋巴细胞增殖缓慢导致适应性免疫相对滞后且免疫应答能效较低,因此鱼类先天性免疫在抵御病原体感染的过程中扮演更为重要的角色[5]. 研究表明,先天性免疫系统主要通过免疫细胞表面的模式识别受体(pattern recognition receptor,PRR)并结合病原体中存在的病原体相关分子模式(pathogen-associated molecular pattern,PAMP),包括微生物的CpG ODN、脂蛋白、dsRNA等,从而引发一系列的免疫反应,以消灭入侵的病原体[6]. 其中,抗原呈递细胞上表达的Toll样受体(Toll-like receptor,TLRs)家族是关键的PRR,在诱导先天性免疫应答以及适应性免疫应答中起着重要作用[3]. 根据不同TLRs识别不同病原相关分子模式,鱼类TLR通常分为两类,第一类包括TLR1、TLR2、TLR4、TLR5和TLR9,主要参与机体对细菌的识别,被称为非病毒型TLRs;第二类包括TLR3、TLR7、TLR8和TLR22,主要参与病毒的识别,被称为病毒型TLRs[7].
到目前为止,鱼类中已经鉴定出大约23种TLRs[8],典型的TLRs蛋白结构包括亮氨酸重复区(LRRs)、跨膜结构域(TM)、胞内Toll/IL-1受体结构域(TIR),其中LRRs区通过与相应的配体结合,使得Toll样受体被激活,进一步通过TIR结构域与接头分子MyD88(髓样分化因子)结合,诱导产生炎症细胞因子抵御病原菌的入侵,TIR区相对保守,与分子信号转导有关[3]. TLRs在鱼类免疫中的作用已被广泛研究,病原体感染后TLRs相关基因表达量会发生不同的变化来适应特定的生存环境. 大西洋鲷(Sparusaurata)被嗜水气单胞菌感染后,TLR2表达发生上调[9];大黄鱼(Larimichthyscrocea)被嗜水气单胞菌感染后,TLR1、TLR3表达上调,TLR2、TLR22表达下调[10];海鱼分枝杆菌也被证实可以诱导斑马鱼TLR9表达上调[11]. 值得注意的是,TLR9与其他非病毒型TLRs不同,该受体只存在于细胞内部,也是识别细菌和病毒DNA中CpG ODN的主要受体,通过与CpG特定的序列结合最终引起免疫器官的功能性成熟,进而产生一系列免疫效应细胞[12],此外,该受体也被认为能与其配体协同进化,因此是研究鱼类抗病原体免疫适应的重要模型. 在非病毒型TLRs中,TLR1似乎也拥有独特的抗病原免疫机制,尽管目前TLR1在大黄鱼、绿河豚(Tetraodonnigroviridis)、斑马鱼等物种中鉴定出来,并且病原感染实验也表明该受体在鱼类病原免疫中发挥着重要作用,但是目前鱼类TLR1的配体还未明确,相关研究表明TLR1可能需要与TLR2形成异源二聚体发挥作用,因此TLR1仍需要更深入的研究[13-14]. 目前国内外对鲈形目TLRs家族研究大多局限在免疫学实验中,然而其分子进化基础仍不完全清楚. 本研究以17种鲈形目物种的TLR1、TLR9为候选基因,尝试通过分析这两个基因分子进化揭示其先天性免疫机制. 研究结果对于鲈形目鱼类的遗传育种以及疾病防治具有一定的指导意义.
1 材料与方法
1.1 TLR1和TLR9序列获得与比对
本研究共选取17种鲈形目物种,涵盖攀鲈科、鲹科、狼鳚科、鲈科、鮨科、花鲈科、鳄冰鱼科、龙科、南极鱼科、石首鱼科、鲷科,并选取斑马鱼作为外群. 除了真鲷以及黑鲷TLR1基因外,本研究所使用物种的TLR1、TLR9基因序列信息均来自NCBI(http://www.ncbi.nlm.nih.gov/)数据库(表1). 进一步,以大西洋鲷TLR1作为参考序列在真鲷(Pagrusmajor)以及黑鲷(Acanthopagrusschlegelii)基因组中进行本地Blast[15]以获取对应的TLR1基因,E-value值设定为1e-5,其中真鲷基因组来自NCBI数据库,黑鲷基因组为江苏省海洋水产研究所与南京师范大学合作测序数据(未发表). 使用Prank[16]软件对每一个基因基于密码子水平进行多序列比对,然后使用Gblocks[17]软件对序列非保守区域进行比对和人工校对.
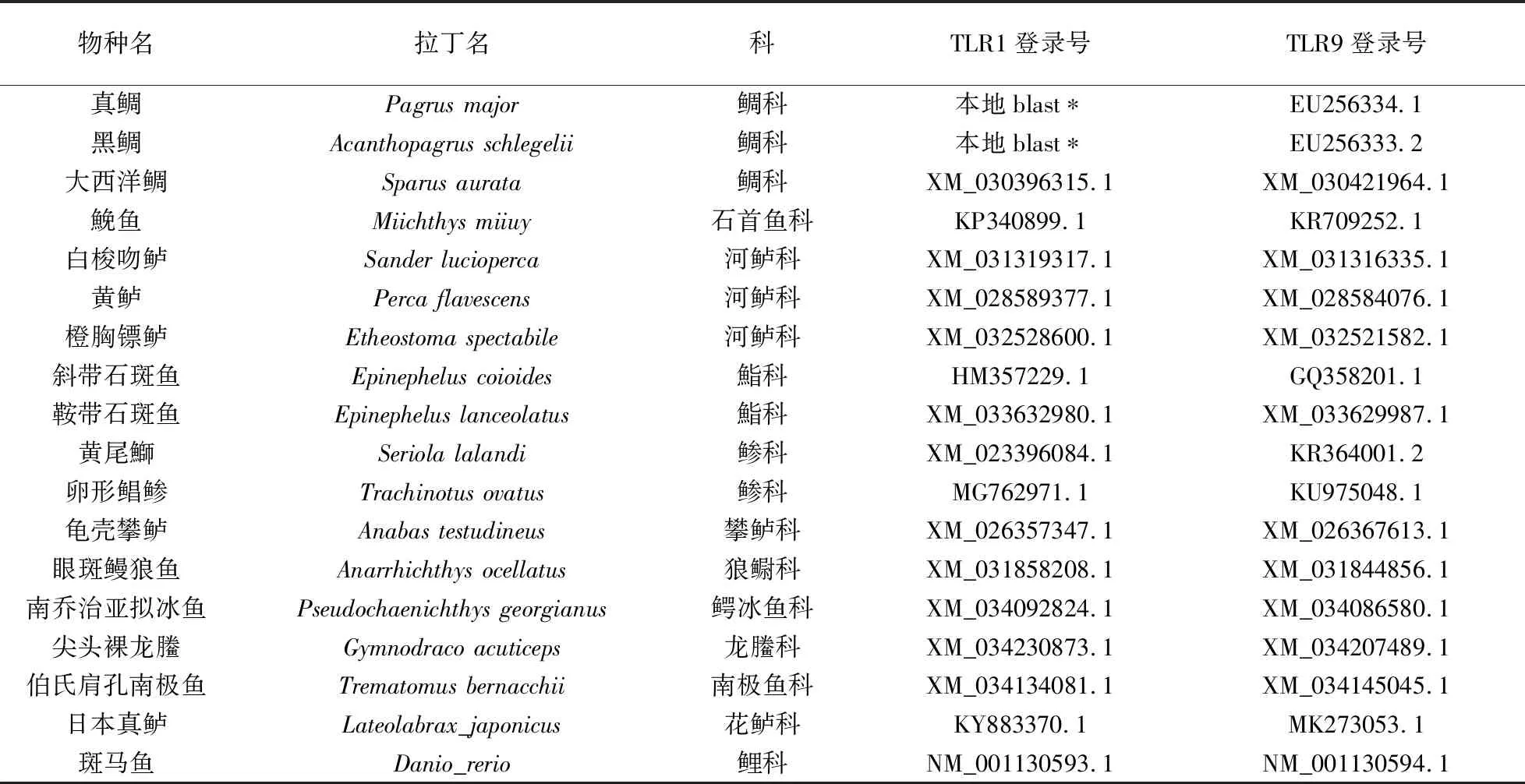
表1 本文研究所选物种基因序列登录号Table 1 Accession numbers of species used in this study
1.2 TLR9和TLR1基因树重建
为探讨TLRs在鲈形目物种内部的系统发生关系,通过Modelgenerator[18]软件选择最佳核苷酸替换模型,使用RAxML[19]软件构建最大似然系统发生树,斑马鱼作为外群,自展1 000次估计节点的支持度(bootstrap value). 另外,为了使得树的结果更加可靠,也使用了Mrbayes[20]软件基于贝叶斯法进行构树,其中后验概率借助马尔科夫链蒙特卡洛方法(markov chain monte carlo,MCMC)运行100000代进行估计.
1.3 分子进化分析
为了探究TLR1和TLR9在鲈形目中的进化模式,本研究使用 PAML4.7(phylogenetic analysis by maximum Likelihood,PAML)软件[21]中CODEML程序对非同义替换率(nonsynonymous,dN)和同义替代率(synonymous,dS)的比值(ω=dN/dS)进行评估.ω值为衡量选择压力的重要指标,其中ω<1、=1和>1分别意味着受纯化选择(Purify selection)、中性选择(Neutral selection)和正选择(Positive selective). 使用目前公认的鲈形目系统发生关系[22]以及TimeTree网站(http://www.timetree.org/)的系统发育树作为PAML分析的输入树(图1).
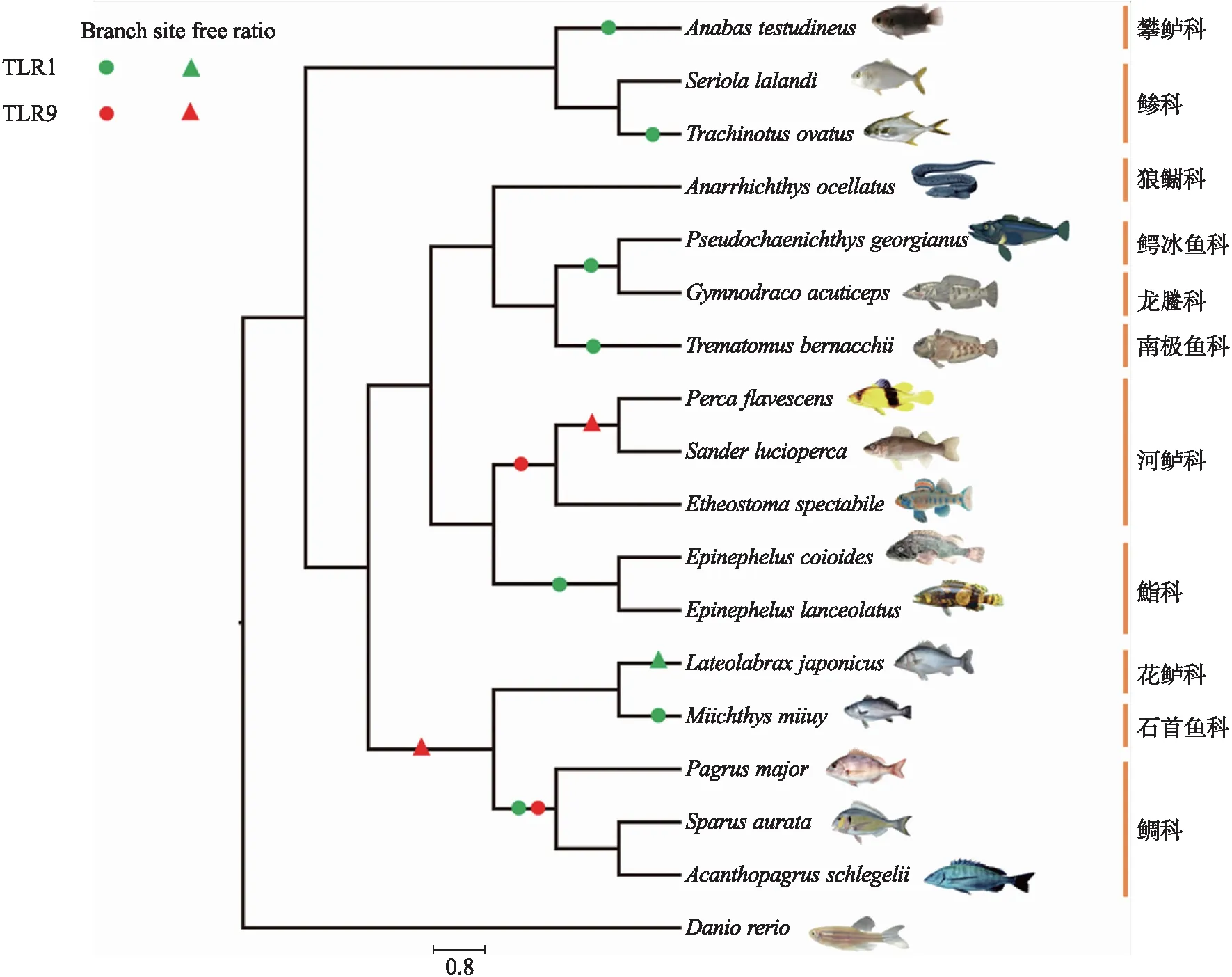
图1 本研究用于PAML分析的输入树. Branch-site、Free ratio结果分别使用圆圈以及三角符号标记Fig.1 The input phylogenetic tree used in PAML analyses in this study. The results of Branch-site and Free ratio models were marked with circles and triangles,respectively
运用基于最大似然法的PAML4.7软件包[21]和Datamonkey[23]进行评估鲈形目物种中TLR1和TLR9两个基因座中受正选择作用的位点. 首先,运用PAML 软件包中的位点模型(Site model)中的两对模型:M1a(中性模型)vs. M2a(正选择模型)、 M8a(中性模型,beta 分布:0<ω0<1和ω1=1)vs. M8(正选择模型;beta分布:0<ω0<1和ω1>1)进行检测. 运用似然比检验(likelihood ratio test,LRT)评估嵌套模型中的最佳模型,并通过2ΔlnL值与自由度之间的卡方分布(Chi-square test,χ2)关系检测上述两个模型的显著性. 当模型通过LRT检验且P值显著时,运用贝叶斯经验贝叶斯法[24](bayes empirical bayes,BEB)检测正选择位点,其中后验概率(posterior probabilities,PP)值大于0.9的位点作为潜在的正选择位点. 另外,基于同义替代的Datamonkey[23]方法进一步用于正选择位点的检测,运用了三种最大似然法,包括固定效应似然法(fixed effects likelihood,FEL)、随机效应似然法(random effects likelihood,REL)、快速无约束贝叶斯估算法(fast unconstrained bayesian approximation,FUBAR). FEL显著性水平设置为0.2,REL贝叶斯显著水平设置为50,FUBAR中后验概率固定阈值0.8.
为了进一步检测正选择是否局限于特定的进化谱系,运用PAML4.7软件包中的自由比率模型(Free-ratio model)和支位点模型(Branch-site model)进行检测. 自由比率模型假设每一支系具有独立的ω值,该模型与零假设——单一比率模型(One-ratio model)即所有支系具有相同的ω值进行比较. 支位点模型(Branch-site model)是最严格的选择压力检测的方法,不仅能够检测受正选择的支系还能检测受正选择的位点. 该模型需要将进化谱系分为前景支(foreground branch,即感兴趣的支系)与背景支(background branch,即其余的支系). 本研究中,将17种鲈形目物种分别标记为前景支,零假设Ma0(中性模型:0<ω0<1,ω1=1和ω2=1)和备择假设Ma(正选择模型:0<ω0<1,ω1>1和ω2≥1)进行比较. 运用LRT检测嵌套模型的显著性,P<0.05时则为模型显著,且所检测的位点PP>0.9时,被认为是正选择位点.
1.4 TLR1和TLR9蛋白三维结构预测
通过MEGA[22]软件将TLR1、TLR9完整核苷酸序列翻译为氨基酸序列,利用SMART网站(http://smart.embl-heidelberg.de/)预测蛋白关键结构域. 蛋白质结构域中氨基酸的变化可能引起蛋白质空间构型的改变,进而影响蛋白质功能. 为了进一步为探究正选择位点在蛋白质三维结构上的空间分布,本研究使用I-TASSER(Iterative Threading ASSEmbly Refinement)网站对黑鲷TLR1和TLR9蛋白三维结构进行预测,并通过EZMOL(http://www.sbg.bio.ic.ac.uk/ezmol/)网站定位正选择位点在蛋白质三维结构上的位置,从而更直观地分析位点对蛋白质生物学功能的影响.
2 结果与讨论
2.1 系统发育树的构建
为了探讨鲈形目TLRs的系统发生关系,首先,基于Modelgenerator[19]软件分析,发现 GTR+GAMMA+I模型为核苷酸替代的最佳模型. 其次,使用RAxML软件构建基因最大似然树(图2). 基因树的拓扑结构显示鲈形目TLRs的基因根据TLR1和TLR9两个基因座聚成了两个基因簇. 但是,基因树的拓扑结构与物种树存在差异,在TLR1物种树中,日本真鲈(Lateolabraxjaponicus)与鮸鱼(Miichthysmiiuy)聚在一起,而在基因树中,鮸鱼与鲷科的大西洋鲷拓扑关系最近. 在TLR9基因树中,除了两个鮨科物种斜带石斑鱼(Epinepheluscoioides)以及鞍带石斑鱼(E.lanceolatus)没有与黄鲈(Percaflavescens)聚在一起,基因树与物种树其他物种亲缘关系基本一致. 同时,贝叶斯树也同样支持了最大似然法构建的基因树,两种方法得到的物种基因树在整体拓扑结构上基本一致(图3). 综上,基因树与物种树的拓扑差异表明在鱼类进化过程中,TLR1、TLR9基因已经发生了独特的进化,以适应不同生境下病原微生物的侵染.

红色和蓝色三角形、五角星分别代表鲈形目TLR1以及TLR9支系. 节点数字表示支持度大于50%图2 最大似然法构建鲈形目TLR1、TLR9系统发育树Fig.2 Reconstruction of the TLR1 and TLR9 phylogenetic tree of Perciformes by the maximum likelihood method

红色和蓝色五角星三角形、五角星分别代表鲈形目TLR1以及TLR9支系. 节点数字表示后验概率图3 贝叶斯法构建鲈形目TLR1、TLR9系统发育树Fig.3 Reconstruction of the TLR1 and TLR9 phylogenetic tree of Perciformes by Bayesian method
2.2 选择压力分析
为了检测TLR1和TLR9两个基因座在鲈形目物种中是否存在正选择位点,首先运用PAML4.7软件中位点模型(M1a vs. M2a、M8a vs. M8)进行检测. LRT结果显示正选择模型M2a和M8均显著优于中性模型M1a和M8a. 在TLR1基因座位中,正选择模型M2a、M8中ω值分别为2.55和2.05,提示TLR1受正选择,并且M2a,M8通过BEB方法分别检测到3个、47个正选择位点的后验概率大于0.9,在TLR9检测结果中,M2a、M8模型分别检测到3、30个正选择位点. 当后验概率提高到0.95时,在TLR1检测结果中,M2a,M8分别筛选到1个、20个正选择位点,而在TLR9中,M2a模型未检测到后验概率大于0.95的位点,M8模型则检测到10个正选择位点. 另外,采用DataMonkey网站中的三种最大似然法(FEL、REL以及FUBAR)进一步对两个基因座位进行选择压力检测. 结果表明,运用三种方法在TLR1基因座位(FEL:28,REL:8,FUBAR:13)以及TLR9基因座位(FEL:24,REL:6,FUBAR:3)共检测53个正选择位点. 综合以上五种最大似然法,TLR1与TLR9中被两种以上方法同时检测到的正选择位点分别为16和8个,提示这些位点为强烈的正选择位点,同时被三种以上方法检测到的正选择位点分别为7个和3个(表2).

表2 PAML和Datamonkey检测到的正选择位点Table 2 Sites under positive selection detected by PAML and Datamonkey
运用单一比率模型(One-ratio model)假设所有支系具有相同的进化速率,结果显示鲈形目物种TLR1和TLR9的ω值分别为0.38与0.36,显著小于1,提示这两个基因整体受到强烈的选择约束. 为了进一步探究正选择是否局限于鲈形目特异的支系,运用自由比率模型(Free-ratio model)进行检测. LRT结果显示嵌套模型(One ratio vs. Free ratio)差异显著(P<0.01),说明自由比率模型显著优于单一比率模型. Free-ratio模型发现TLR1在日本真鲈显著受正选择;而TLR9基因座位中则检测到多个正选择支系,例如黄鲈与白梭吻鲈(Sanderlucioperca)最近共同祖先支,日本真鲈、鮸鱼、真鲷、大西洋鲷以及黑鲷的最近共同祖先支(图2、表3). Free ratio模型检测到一些支系中存在正选择信号(ω>1),据报道这些支系长期受到病原微生物的侵袭,比如鲷科支系已被报道经常受到虹彩病毒侵袭,引起组织细胞肿大坏死[25].
运用最严格的支位点模型(Branch-site model)检测选择压力,结果提示在TLR1基因座位中,正选择分别发生在龟壳攀鲈(Anabastestudineus)、鮸鱼、卵形鲳鲹(Trachinotusovatus)、伯氏肩孔南极鱼(Trematomusbernacchii)以及南乔治亚拟冰鱼(Pseudochaenichthysgeorgianus)、尖头裸龙(Gymnodracoacuticeps)最近共同祖先支,斜带石斑鱼和鞍带石斑鱼最近共同祖先支,真鲷、黑鲷及大西洋鲷最近共同祖先支;在TLR9中,黄鲈、白梭吻鲈以及橙胸镖鲈(Etheostomaspectabile)最近共同祖先支,真鲷、黑鲷及大西洋鲷最近共同祖先支也分别检测到正选择位点(表4).

表3 基因位点模型和分支模型检测结果Table 3 The result for site model and branch model(PAML)tests of genes
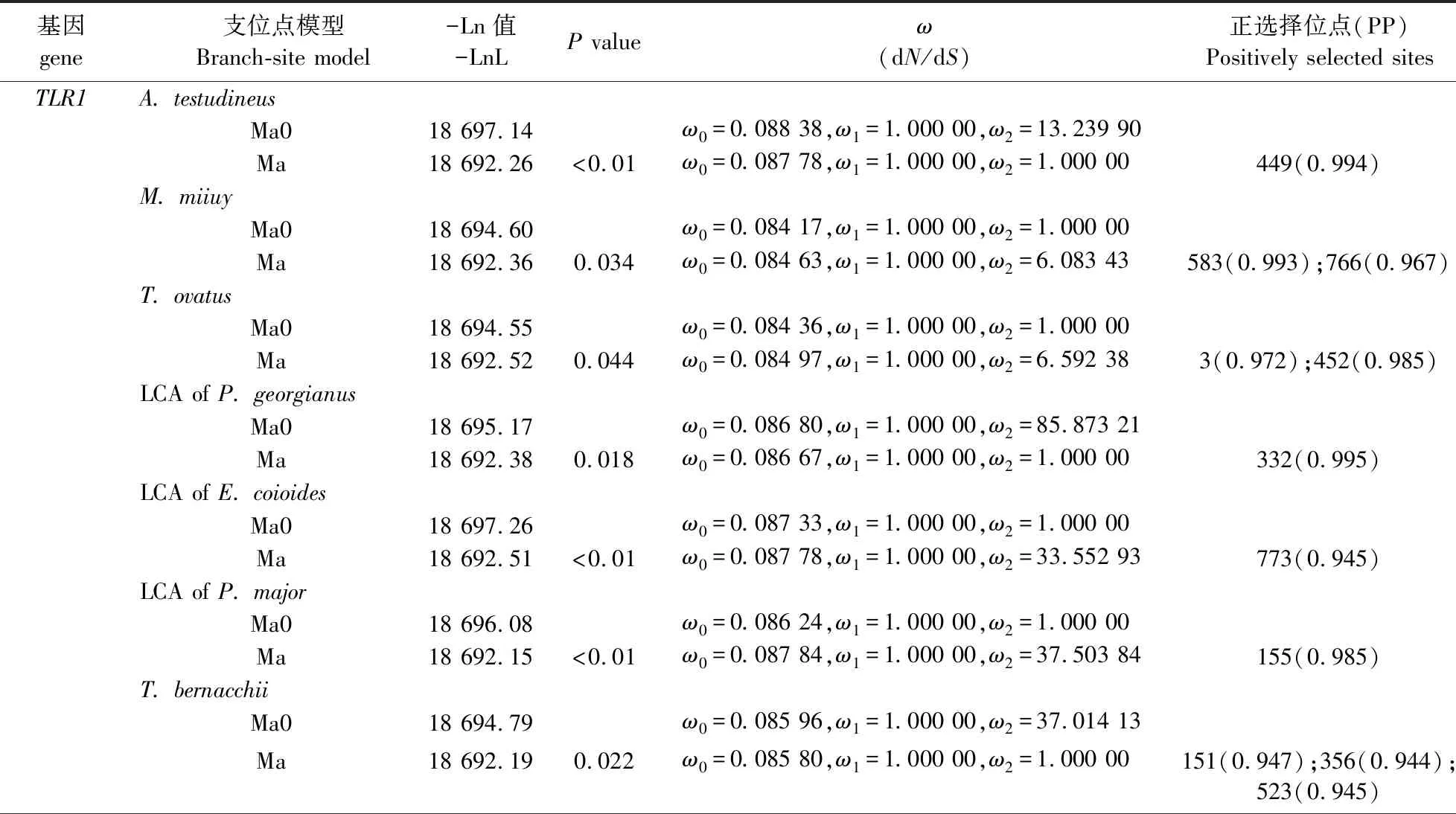
表4 TLR1和TLR9基因支位点模型检测结果Table 4 The result for Branch-site model test of genes

续表4 Table 4 continued
2.3 TLR1、TLR9结构预测以及正选择位点标注在三维结构中
本研究选取了亲缘关系较远的大西洋鲷(鲷科)、伯氏肩孔南极鱼(南极鱼科)、龟壳攀鲈(攀鲈科)三个物种为代表进行氨基酸序列分析. 利用SMART网站分别预测TLR1、TLR9蛋白结构域并通过IBS[26]软件对结构域进行可视化(图4). 龟壳攀鲈、大西洋鲷、伯氏肩孔南极鱼三个物种中,TLR1编码序列长度相似,分别为798、802、802个氨基酸,其中大西洋鲷具有最多的亮氨酸重复区(13个),但值得注意的是,此物种未检测到羧基端亮氨酸重复区;伯氏肩孔南极鱼亮氨酸重复区数量最少(4个)并且未检测到跨膜区. TLR9在三个物种中相对更为保守,都含有典型的TLRs蛋白结构域(LRR、TM、TIR),其中亮氨酸重复区数量分别为12、13、15个. 为了进一步探究正选择位点对蛋白质功能的影响,将这些位点标注在蛋白三维结构中,首先利用I-TASSER网站预测了黑鲷蛋白三维结构模型并通过EZMOL在线网站将同时被两种最大似然法鉴定到的正选择位点(TLR1:16和TLR9:8)标注到三维模型中(图5),发现58%的正选择位点位于蛋白质重要功能域,且86%强烈的正选择位点位于病原体识别区域亮氨酸重复区(LRRs).

SP:信号肽;LRR:亮氨酸重复区;TM:跨膜区;LRRCT:羧基端亮氨酸重复区;TIR:Toll/IL-1受体结构域.图4 不同物种TLR1、TLR9蛋白结构比较Fig.4 Comparison of the TLR1、TLR9 domain structures among various species
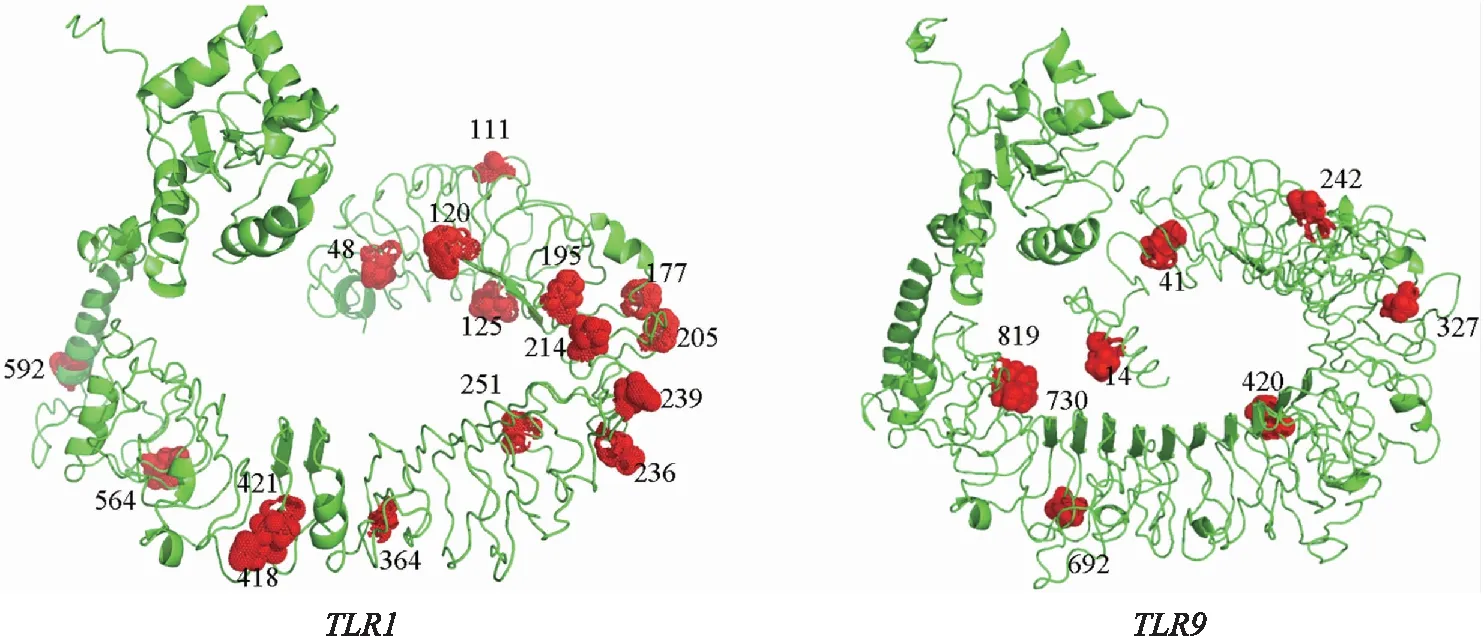
图5 基因正选择位点在蛋白三维结构中的标注Fig.5 Annotation of the positive selection sites of genes in the three-dimensional structure of the protein
3 结论
TLRs是进化保守的蛋白质,传统上受到强烈的功能约束. 然而,最近的研究已经在一些脊椎动物群体中发现正选择的证据,如鸟类中TLR3、4、5和15基因、啮齿动物的TLR4基因等[27-29]. 相对于恒温哺乳类、鸟类物种,生活在水体中的鱼类因没有自我调节体温的机制,当面临病原微生物入侵时,低温诱导的淋巴细胞的增值减少将导致适应性免疫相对滞后且免疫应答能效较低,因此对栖息于病原体较多的水环境鱼类而言,先天性免疫在抵御病原体侵染的过程中扮演更为重要的角色[28]. 作为Toll样受体基因家族中的重要成员,TLR1、TLR9分别可以识别三酰基脂肽、细菌与病毒非甲基化的CpG寡核苷酸序列,尽管在鱼类免疫识别应答方面的功能已被广泛的研究,但是基因的分子进化基础尚不清楚. 目前,已经报道了鲈形目物种容易受到不同的病原微生物的感染,包括真鲷虹彩病毒(RSIV)、鲈鱼虹彩病毒(WVIV)、淋巴囊肿病毒(LCDV)、红细胞坏死病毒(VENV)、传染性胰腺坏死病病毒(IPNV)、流行性造血器官坏死病毒(EHNV)、神经坏死症病毒(NNV)等[25,30]. 本研究通过分析鲈形目物种先天免疫相关基因TLR1、TLR9的进化模式,发现物种普遍受到显著的正选择. 在位点模型中,TLR1与TLR9中被两种以上方法检测到的正选择位点分别为18、6个,且86%位点处于LRR区. 已报道LRR结构域在识别病原体组分中发挥核心作用,且LRR和LRR_CT区域还参与病原体刺激后的信号转导过程[31]. 该区检测到更多正选择位点可能有利于物种在进化过程中识别不同的配体进而有效的抵抗病原微生物. 另外,相比于LRR区,TIR区具有更高的保守性,并且在信号转导过程中发挥重要作用. 鲈形目物种在TIR结构域未检测到正选择位点,提示该区域可能受到强烈的功能约束.
特别重要的是,也在鲈形目一些支系中检测到特有的正选择信号,并且检测到的正选择位点与位点模型所检测到的位点不同,这些支系可能经历了更多的选择来应对不同病原体的感染. 两个基因在鲷科的最近共同祖先支均检测到正选择信号,之前研究也证明鲷科物种经常感染淋巴囊肿病、病毒性表皮坏死病等[25]. 石斑鱼属是环境适应能力较强的暖水性鱼类,尤其是斜带石斑鱼,生活温度可以在11~41 ℃之间. 目前,病毒性神经坏死症是石斑鱼流行病之一[32],研究也在两个石斑鱼属物种(斜带石斑鱼、鞍带石斑鱼)的祖先支检测到TLR1基因正选择的信号,可能与其独特的环境适应能力有关. 另外,河鲈科物种被列为病毒性出血性败血症的易感物种[33]. 作为全球有鳍鱼类致病性最强的病毒性疾病之一,宿主致死率可高达90%,在河鲈科物种的祖先支中也检测到TLR9基因存在正选择信号. 这些物种检测到显著的正选择信号,表明随着时间的推移,物种抗病毒免疫反应可能已经发生了进化(图1,表4),提示鲈形目物种TLR1、TLR9的分子进化可能是由环境中的病原微生物介导的选择压力所驱动的,从而具有较强的抵御病原微生物侵袭的能力.
另外,两个基因受正选择的比例也明显不同,在TLR1中检测到更多的正选择信号,并且系统发育树也表明TLR1基因树与物种树相比,发生了更多的拓扑结构变化,表明鱼类在适应病原微生物的过程中,TLR1、TLR9基因受到不同的选择机制且TLR1进化更为明显. 本研究也发现58%的正选择位点位于蛋白质重要功能域,并且大多数位于病原体识别区域亮氨酸重复区(LRRs),该区域的进化有利于识别不同病原体PAMP,从而在抗病原免疫过程中发挥重要作用(图4). 以上研究结果从进化的角度初步揭示了鲈形目物种先天性免疫基因TLR1、TLR9与病原体的相关性,为物种的疾病防治与预防提供理论依据.
猜你喜欢
科学(2020年3期)2020-11-26 08:18:22
科学(2020年3期)2020-11-26 08:18:22
——以云南墨江自治县为例
贵州民族研究(2019年9期)2019-10-24 03:27:36
广州大学学报(自然科学版)(2019年1期)2019-05-07 01:33:26
神州民俗(2018年9期)2018-11-21 11:10:02
现代园艺(2017年13期)2018-01-19 02:27:58
中学生数理化·八年级物理人教版(2017年6期)2017-11-09 06:00:44
流行色(2017年1期)2017-05-31 19:18:01
新西部·中旬刊(2016年5期)2016-06-08 10:25:35
天津科技大学学报(2016年1期)2016-02-28 16:59:45
