
Necroptosis plays a crucial role in the exacerbation of retinal injury after blunt ocular trauma
2022-09-16 04:19YuHuanXiuQuanWuTaoChenYaNanDouBoJiaXinHeDongYuWeiZhouFeiFeiFei
中国神经再生研究(英文版) 2023年4期
Yu Huan , Xiu-Quan Wu , Tao Chen , Ya-Nan Dou Bo Jia Xin He Dong-Yu Wei Zhou Fei , Fei Fei
Abstract Retinal injury after blunt ocular trauma may directly affect prognosis and lead to vision loss.To investigate the pathological changes and molecular mechanisms involved in retinal injury after blunt ocular trauma, we established a weight drop injury model of blunt ocular trauma in male Beagle dogs.Hematoxylin-eosin staining, immunofluorescence staining, western blotting, and TUNEL assays were performed to investigate retinal injury within 14 days after blunt ocular trauma.Compared with the control group, the thicknesses of the inner and outer nuclear layers, as well as the number of retinal ganglion cells, gradually decreased within 14 days after injury.The number of bipolar cells in the inner nuclear layer began to decrease 1 day after injury, while the numbers of cholinergic and amacrine cells in the inner nuclear layer did not decrease until 7 days after injury.Moreover, retinal cell necroptosis increased with time after injury; it progressed from the ganglion cell layer to the outer nuclear layer.Visual electrophysiological findings indicated that visual impairment began on the first day after injury and worsened over time.Additionally, blunt ocular trauma induced nerve regeneration and Müller glial hyperplasia; it also resulted in the recruitment of microglia to the retina and polarization of those microglia to the M1 phenotype.These findings suggest that necroptosis plays an important role in exacerbating retinal injury after blunt ocular trauma via gliosis and neuroinflammation.Such a role has important implications for the development of therapeutic strategies.
Key Words: Beagle dogs; blunt ocular trauma; gliosis; M1 microglia; Müller cells; necroptosis; neuroinflammation; retinal ganglion cells; retinal injury; weight drop injury
Introduction
Ocular trauma is defined as physical and functional damage to the eye and its appendages as a result of external factors (Pieramici et al., 1997, 2003).As one of the leading causes of vision loss, ocular trauma is responsible for monocular vision loss in approximately 20 million people worldwide; closed mechanical ocular trauma (i.e., blunt ocular trauma) is the most important type (Razeghinejad et al., 2020; Shah et al., 2020).Blunt ocular trauma also frequently occurs in modern wars and terrorist attacks (Shah et al., 2020; Li et al., 2021b).Retinal injury after ocular trauma greatly reduces the therapeutic effect and places substantial economic burdens on society and affected families (Mammadova et al., 2017).Therefore, it is important to explore the pathological changes and molecular mechanisms involved in retinal injury after blunt ocular trauma.
Necroptosis is a form of cell death identified in the past 10 years; it is characterized by the production of reactive oxygen species, a large number of pro-inflammatory factors, and the promotion of DNA degradation (Galluzzi et al., 2017; Yuan et al., 2019; Yan et al., 2021, 2022).Necroptosis is identified through detection of the necrosome, a complex composed of receptorinteracting protein kinase (Rip)1, Rip3, and mixed lineage kinase domainlike protein (MLKL) (Galluzzi et al., 2017; Yuan et al., 2019).Necroptosis has an important role in retinal injury, and the application of its inhibitor necrostatin-1 provides robust protection of cells such as retinal ganglion cells(RGCs) (Dvoriantchikova et al., 2014; Thomas et al., 2020; Gao et al., 2021;Yu et al., 2021).Wu et al.(2022) reported that retinal cell apoptosis also contributes to retinal injury through a mechanism that involves the Homer1 protein family.Glia with immune properties are present in the retina; of these, Müller cells and microglia are most important.Müller cells are retinaspecific cells that can differentiate and regenerate into neurons under certain conditions; however, abnormal activation of Müller cells can result in glial hyperplasia (Devoldere et al., 2019).Microglia are ubiquitous immune cells in the central nervous system that can undergo M1/M2 polarization in response to external stimuli; they have pro-inflammatory and anti-inflammatory roles (Li et al., 2015; Silverman and Wong, 2018; Rathnasamy et al., 2019).
The pathological changes and molecular mechanisms involved in retinal injury after blunt ocular trauma have not been fully characterized.Published studies have mainly focused on pathology and mechanisms in the hyper-acute phase(5-48 hours) after injury (Bricker-Anthony et al., 2014; Thomas et al., 2018,2019).There is a need to characterize the retinal injury that occurs in later phases.
Currently, there is no effective treatment for retinal injury after blunt ocular trauma, owing to undefined pathogenesis.Although mice and rats are themain model animals of retinal injury, there are differences between rodent and human retinas (Jeon et al., 1998; Jacobs et al., 2004).Large animals,including dogs, are considered to be model animals closer to humans, and have been widely used in preclinical researches (Matsunaga et al., 2019).Actually, the pathological changes of mice might not be similar with those of dogs.A study revealed that AP-1 was the critical player in the retinal celldeath signal in rodents rather than dog microglia s (Gu et al., 2009), and another study indicated that there were distinctive characteristics existing in dog retina (Genini et al., 2014), suggesting that the mechanism of canine retinal injury needs to be further investigated.Here, we used a weight drop injury model of blunt ocular trauma in Beagle dogs to investigate the pathological changes and related mechanisms involved in retinal injury within 14 days after blunt ocular trauma.
Methods
Animals
All animal procedures were performed in accordance with the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research; the study protocol was approved by the Animal Ethics Committee of Air Force Medical University (approval No.IACUC-20210421) on April 21, 2021.Thirty-two male Beagle dogs (22-24 months old, 17-20 kg) from Dilepu Biomedical Co., Ltd.(Xi’an, China) (license No.SCXK 2019-002) were included in this study and housed in a clean condition.In this study, only male Beagle dogs were used to avoid potential sex differences because such differences have been observed in human and mouse retinas (Bessinis et al., 2013; Trotta et al., 2021).The dogs were housed at 25 ± 1°C with a relative humidity of 40-70% and a 12-hour light/dark cycle; they were given food and waterad libitum
.The experimental design is shown in Figure 1.
Figure 1|Flowchart of experimental procedures.
Experimental groups and establishment of the weight drop injury model
Beagle dogs were randomized into control (n
= 8) and trauma (n
= 24) groups.Dogs in the trauma group were further stratified into 1 (n
= 6), 3 (n
= 6),7 (n
= 6), and 14 days (n
= 6) after injury; dogs in the control group were anesthetized but not injured.The weight drop injury model was established in accordance with a published method (Blanch et al., 2012): dogs in both control and trauma groups were anesthetized with 2-3% isoflurane (Hengrui, Lianyungang, China) in oxygen and placed on a sterile console.In each Beagle dog, one eye was randomly selected.To establish the weight drop injury model, a sterile steel ball (300 g) with a diameter of 4 cm was vertically dropped along a plastic tube with a length of 1 m onto each dog’s lateral sclera (Blanch et al., 2012); Beagle dogs in the control group did not experience trauma.Subsequently, in the trauma group, half of the injured eyes were randomly selected for incubation in paraformaldehyde (4%), while the remaining injured eyes were stored at-80°C until retinal tissue separation.
Hematoxylin-eosin staining
Hematoxylin-eosin (HE) staining was performed to explore damage in the outer nuclear layer (ONL), inner nuclear layer (INL), and ganglion cell layer (GCL).Prior to histological analysis, eyes were immersed in 4%paraformaldehyde for 24 hours at 4°C.Paraffin-embedded sections (4 μm thickness) were prepared and subjected to HE staining.The sections were placed in aqueous hematoxylin solution for 10 minutes.Then, they were incubated in acid water and ammonia water for 30 seconds each.Next, they were washed with running water for 1 hour, then dehydrated in 70% and 90%alcohol for 10 minutes each.Finally, they were treated with an alcoholic eosin staining solution for 2-3 minutes.Light microscopy images were obtained using an FV10i microscope (Olympus, Tokyo, Japan).The central area of the sagittal plane was identified in each retina.Retinal sections within 3000 μm from the optic nerve were analyzed; four fields per retina were studied.The thicknesses of ONL and INL were then measured.
Immunohistochemistry
Immunohistochemistry was performed to investigate various subtypes of cells in the INL and among immune cells.Eyes were immersed in 4%paraformaldehyde phosphate buffer (pH 7.4) for 24 hours at 4°C.Serial sections (4 μm thickness) were prepared.For immunostaining, the sections were blocked with 0.01 M phosphate-buffered saline (PBS) containing 0.3% Triton X-100 and 3% bovine serum albumin for 1 hour.Sections were then incubated at 4°C overnight with one or more of the following primary antibodies [all diluted 1:100 by a fresh blocking buffer (0.01 M PBS containing 0.3% Triton X-100 and 3% bovine serum albumin)]: synaptophysin (Syn;rabbit; Aviva Systems Biology, San Diego, CA, USA; Cat# ARP45435, RRID:AB_2048301), Ki67 (rabbit; Proteintech, Wuhan, China; Cat# 27309-1-AP,RRID: AB_2756525), glutamine synthetase (GS; mouse; Proteintech; Cat#66323-2-Ig, RRID: AB_2881704), CD45 (rabbit; Proteintech; Cat# 20103-1-AP, RRID: AB_2716813), sex determining region Y-box 2 (SOX2) (rabbit;Novus, Littleton, CO, USA; Cat# NB110-37235, RRID: AB_792070), protein kinase C (PKC; mouse; Enzo, New York, NY, USA; Cat# ADI-KAM-PK020, RRID:AB_10998587), inducible nitric oxide synthase 2 (iNOS) (mouse; Santa Cruz Biotechnology, Dallas, TX, USA; Cat# sc-7271, RRID: AB_2891105), glial fibrillary acidic protein (GFAP; mouse; Millipore, Burlington, MA, USA; Cat#MAB3402, RRID: AB_94844), calretinin (rabbit; Abcam, Cambridge, UK; Cat#ab702, RRID: AB_305702), choline acetyltransferase (ChAT; rabbit; Abcam;Cat# ab6168, RRID: AB_2244866), ionized calcium binding adapter molecule 1 (Iba1) (rabbit; Abcam; Cat# ab178847, RRID: AB_2832244), Iba1 (mouse;Abcam; Cat# ab15690, RRID: AB_2224403), calbindin-D (rabbit; Sigma, St.Louis, MO, USA; Cat# c2724, RRID: AB_258818), or CD16/32 (rabbit; Abcam;Cat# ab223200).After incubation with primary antibodies, sections were washed with PBS for three times, then incubated for 2 hours at 20-25°C in the dark with the appropriate secondary antibodies (all diluted 1:200 by PBS):Alexa Fluor488 (donkey anti-rabbit; Thermo Fisher Scientific, Waltham,MA, USA; Cat# A-21206, RRID: AB_2535792), Alexa Fluor™ 488 (donkey anti-mouse; Thermo Fisher Scientific; Cat# A-21202, RRID: AB_141607), Alexa Fluor™ 594 (donkey anti-rabbit; Thermo Fisher Scientific; Cat# A-21207,RRID: AB_141637), or Alexa Fluor594 (donkey anti-mouse; Thermo Fisher Scientific; Cat# A-21203, RRID: AB_141633).Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) (1:5000, Sigma; Cat# D9542).All immunostained sections were photographed under a fluorescent microscope(FV10i) with identical settings and cell numbers were quantified using ImageJ 1.8.0 software (National Institutes of Health, Bethesda, MD, USA) (Schneider et al., 2012).The central area of the sagittal plane was identified in each retina.Retinal sections within 3000 μm from the optic nerve were analyzed;four fields per retina were studied.
TdT-mediated dUTP nick end labelling assays
TdT-mediated dUTP nick end labelling (TUNEL) assays were used to investigate retinal cell death.Retinal sections were analyzed using a terminal deoxynucleotidyl transferase enzyme kit (40306ES50, Yeasen, Shanghai,China), then fluorescently labeled with 2′-deoxyuridine 5′-triphosphate at 37°C for 1 hour.Nuclei were stained with DAPI.The numbers of TUNEL-positive cells were determined by fluorescence microscopy.
Western blotting assay
Western blotting assays were performed to investigate necroptosis-related protein expression.Proteins were prepared by direct lysis of retinal tissues using radio immunoprecipitation assay buffer (Sigma; Cat# R0278).For each sample, 30 μg of protein was separated by 10-15% sodium dodecyl sulfatepolyacrylamide gel electrophoresis, then electroblotted onto a polyvinylidene difluoride membrane.Each membrane was blocked with PBS (pH 7.4)containing 10% bovine serum albumin for 1 hour, then incubated overnight at 4°C with one of the following primary antibodies (all diluted 1:1000 by antibody diluent (WB100D, NCM Biotech, Suzhou, China)): phospho-Rip3(p-Rip3; rabbit; Cell Signaling Technology, Danvers, MA, USA; Cat# 91702),Rip3 (rabbit; Cell Signaling Technology; Cat# 95702), MLKL (rat; Millipore;Cat# MABC604, RRID: AB_2820284), phospho-MLKL (p-MLKL; mouse;Millipore; Cat# MABC1158), or β-actin (mouse; Proteintech; Cat# 66009-1-Ig, RRID: AB_2687938).After the membrane had been washed with PBS for three times, it was incubated for 2 hours at 20-25°C with one of the following horseradish peroxidase-conjugated secondary antibodies (all diluted 1:3000 by antibody diluent (WB100D)): anti-rabbit (Cell Signaling Technology; Cat#7074, RRID: AB_2099233), anti-mouse (Cell Signaling Technology; Cat# 7076,RRID: AB_330924), or anti-rat (Cell Signaling Technology; Cat# 7077, RRID:AB_2099233).Finally, images were captured with the Molecular Imager System (Tanon 5200, Shanghai, China) and the optical densities of protein bands were quantified using ImageJ software.
Enzyme-linked immunosorbent assay
To explore the expression patterns of inflammatory factors, retinal tissues were collected.Interleukin-1β (IL-1β) and tumor necrosis factor-a (TNF-α)levels in the retina were detected using commercially available enzymelinked immunosorbent assay kits (Proteintech) in accordance with the manufacturer’s instructions.
Full-field electroretinogram recording
Visual performance in the experimental dogs was measured as previously described (Kraszewska et al., 2016).Briefly, Beagle dogs were subjected to 30 minutes of dark adaptation, then anesthetized with isoflurane prior to full-field electroretinogram (ERG) assessment.A 0.5% tropicamide solution(Shenyang Xingji Corporation, Shenyang, China) was used for pupil dilation;oxybuprocaine hydrochloride eye drops (Santen Pharmaceutical Co., Ltd.,Osaka, Japan) were used for corneal anesthesia.The center of the cornea was attached with an ERG-Jet corneal electrode (Fabrinal, La Chaux-de-Fonds,Switzerland) served as the active electrode, while the reference electrode and the ground electrode were both circular electrodes and were placed at the base of the ear and the middle of the back, respectively.Dim red light was used during all of the above procedures to maximize retinal sensitivity.ERG responses were recorded by the RETeval device (LKC Technologies,Gaithersburg, MD, USA).Upon completion of the ERG recording, levofloxacin eye drops (Tianlong, Suzhou, China) were administered to prevent infection.
Flash visual evoked potential
Visual electrophysiology was performed in accordance with a previously reported protocol (Strain et al., 1990; Ito et al., 2015).After ERG recording,the flash visual evoked potential (F-VEP) was recorded between Oz (midline of the nuchal crest, positive electrode) and Fpz (midline, just caudal to the eyes, negative electrode), with ground at the middle of the back.The stimulus was a light-emitting diode flash intensity of 3 cd/m·s with 0.99 Hz frequency,generated by the RETeval device (LKC Technologies).
Statistical analysis
A preliminary HE staining experiment revealed that the numbers of RGCs differed between the control group (mean = 0.903, standard deviation = 0.084)and trauma group (mean = 0.508, standard deviation = 0.080).We used these data to calculate the sample size in PASS15 software (NCSS, Kaysville, UT, USA),with power = 0.9 and α = 0.05.Evaluators were blinded to group assignments.Data were expressed as the mean ± standard error of the mean (SEM) of at least three independent experiments.Comparisons between groups were conducted using Student’st
-test in GraphPad Prism software (version 8.0.1,GraphPad, San Diego, CA, USA).Power analyses oft
-tests were performed in PASS 15 software (NCSS) and the results are shown in Additional Table 1.Results
Retinal injury after blunt ocular trauma
HE staining results showed that blunt ocular trauma induced retinal degeneration, rather than retinal detachment (Figure 2).The relative thicknesses of the ONL, INL, and GCL were calculated; the number of cells in the ONL, INL and GCL were compared between the injury and control groups(allP
< 0.01).The relative number of RGCs sharply decreased by nearly 50%on the 14day after injury, compared with the control group.The numbers of cells in the INL and ONL layers began to decrease on the seventh day after injury (P
< 0.01).These findings suggest that blunt ocular trauma can lead to retinal injury and have an impact on ocular health.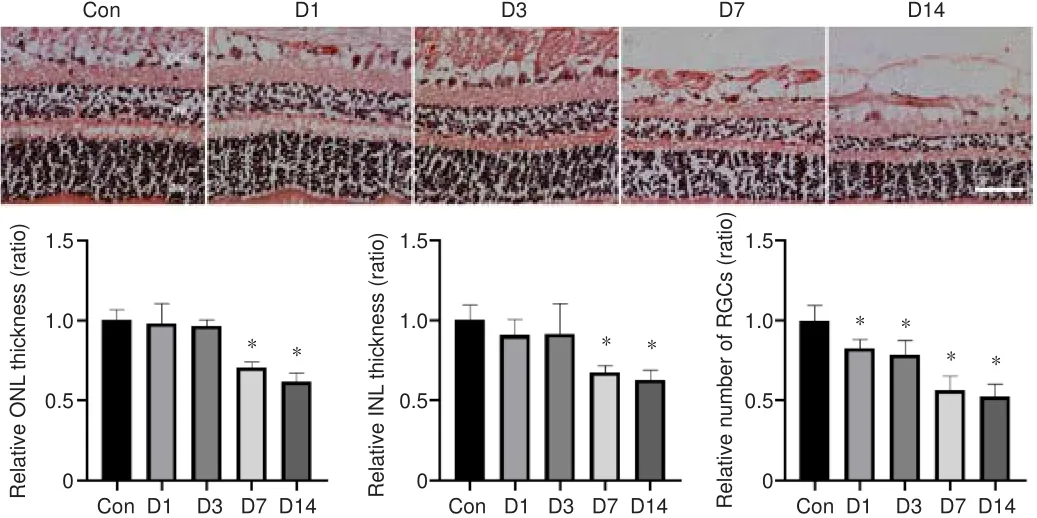
Figure 2| HE staining assessment of retinal injury after blunt ocular trauma.
Damage to the ONL and INL worsens on the seventh day after blunt ocular trauma
Various types of retinal cells responded differently to blunt ocular trauma in this study; the thicknesses of the ONL and INL decreased after injury.Rod and cone photoreceptor cells, which are the main components of the ONL, exhibited injury until the seventh day after blunt ocular trauma;the relative thickness of the ONL decreased over time (Figure 2).Next,immunofluorescence staining was performed to investigate the effects of blunt ocular trauma on horizontal, bipolar, Müller, and amacrine cells in the INL.Staining for ChAT (a marker of cholinergic cells) revealed that the number of cholinergic cells (a subset of amacrine cells) began to decline at 7 days after injury (P
< 0.05; Figure 3A and B).The number of calretinin-positive cells (i.e., amacrine cells (Keilhoff et al., 2021)) also decreased at 7 days after injury (P
< 0.01; Figure 3C and D).The number of calbindin-D-positive cells(i.e., horizontal cells) was unchanged after blunt ocular trauma (P
> 0.05;Figure 3E and F); the relative number of calbindin-D-positive cells also did not change.Nevertheless, PKCα-positive cells (i.e., bipolar cells) appeared to be vulnerable to blunt ocular trauma, such that the number of bipolar cells significantly decreased on the first day after injury (P
< 0.05; Figure 3G and H).Intersections of bipolar cells and synaptophysin-positive photoreceptor ribbon synapses are involved in visual transduction.We found that the connections between bipolar cells and photoreceptor cells were reduced on the seventh day after injury (P
< 0.01; Figure 3G and I).Overall, these results suggest that bipolar cells were affected by blunt ocular trauma in a manner similar to rods and cones; with the exception of horizontal cells, all cells in the INL exhibited injury beginning at 7 days after blunt ocular trauma.Blunt ocular trauma induces retinal cell necroptosis
TUNEL assays showed that retinal cell death occurred on the first day after injury in the INL and ONL (bothP
< 0.01; Figure 4A and B), with dramatic increases in the number of TUNEL-positive cells.The number of TUNEL-positive cells in the INL increased over time, while the number of TUNEL-positive cells in the ONL was sharply increased at 14 days after injury (Figure 4A and B).The progression of retinal injury progressed from the GCL to the ONL (Figure 4A), suggesting that some detrimental factors such as ischemia,oxidative stress and inflammation, might be involved in the process of retinal injury and exacerbation of retinopathy.To investigate the role of necroptosis in blunt ocular trauma, we examined necroptosis-related proteins, such as p-Rip3, Rip3, p-MLKL, and MLKL.We found that the ratios of p-Rip3/Rip3 and p-MLKL/MLKL both increased over time (bothP
< 0.01; Figure 4C), suggesting that necroptosis plays an important role in the exacerbation of retinal injury.Müller cell regeneration and gliosis occur after blunt ocular trauma
Double immunofluorescence staining of Ki67 and GS showed that the number of Ki67-positive Müller cells increased beginning on the first day after trauma and reached a peak on the third day after blunt ocular trauma (bothP
< 0.01;Figure 5A and B).SOX2 and GS staining showed that the number of SOX2-positive Müller cells increased beginning on the first day after blunt ocular trauma (bothP
< 0.01; Figure 5C and D); the relative number of Müller cells increased in a similar manner.Additionally, the number of GFAP-positive cells increased on the seventh day after injury (P
< 0.01), suggesting enhanced Müller cell gliosis (Figure 5E and F).Thus, Müller cell gliosis was activated after Müller cell regeneration.Inflammation and microglial polarization occur after blunt ocular trauma
CD45 staining was performed to assess the inflammatory response; the relative number of CD45-positive cells in the injury group increased overtime compared with that in the control group (bothP
< 0.05; Figure 6A and B).To investigate the role and characteristics of spatiotemporal changes in microglia after blunt ocular trauma, we performed immunofluorescence staining of microglia in the retina.We found that microglia were activated and recruited into the injured retina (Figure 6C-F), such that the relative number of Iba1-positive cells in the retina was elevated (P
< 0.01).CD16/32 and iNOS are cellular markers of M1 microglia, which are presumed to secrete pro-inflammatory cytokines and promote neuroinflammation (Li et al., 2021a).The number of M1 microglia increased on the seventh day after injury (bothP
< 0.01; Figure 6C-F); this was accompanied by increased levels of the pro-inflammatory cytokines IL-1β and TNF-α after injury (bothP
< 0.01; Figure 6G).Moreover, the activation and polarization of microglia followed the pattern of progression from the GCL to the INL and ONL (Figure 6C-F), suggesting that the progression of retinal cell death is associated with microglial activation.ERG and F-VEP findings indicate impaired retinal function after blunt ocular trauma
ERG and F-VEP assessments were performed for objective analysis of visual function.The b-wave amplitude in the dark-adapted 3.0 ERG decreased (P
<0.05), indicating that retinal function was impaired after blunt ocular trauma(Figure 7A and B).The total oscillatory potential amplitude decreased (P
<0.05), suggesting the impairment of retinal microcirculation after blunt ocular trauma (Figure 7A and B).The b-wave amplitude in 30 Hz flicker ERG was unchanged (P
> 0.05; Figure 7A and B).Additionally, F-VEP assessments were conducted to investigate the visual pathway.Decreases in the total amplitudes of P1 and P2 indicated that blunt ocular trauma caused vision impairment in Beagle dogs (P
< 0.05), although the peak times of P1 and P2 were unaffected(P
> 0.05; Figure 7A and B).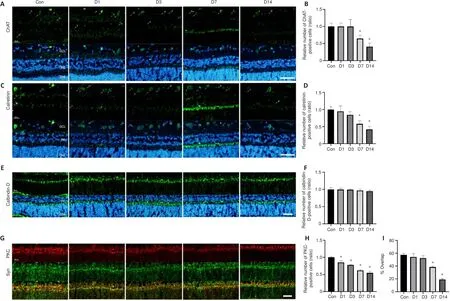
Figure 3| Damage to the ONL and INL worsens on the seventh day after blunt ocular trauma.

Figure 4| Blunt ocular trauma induces retinal cell necroptosis.
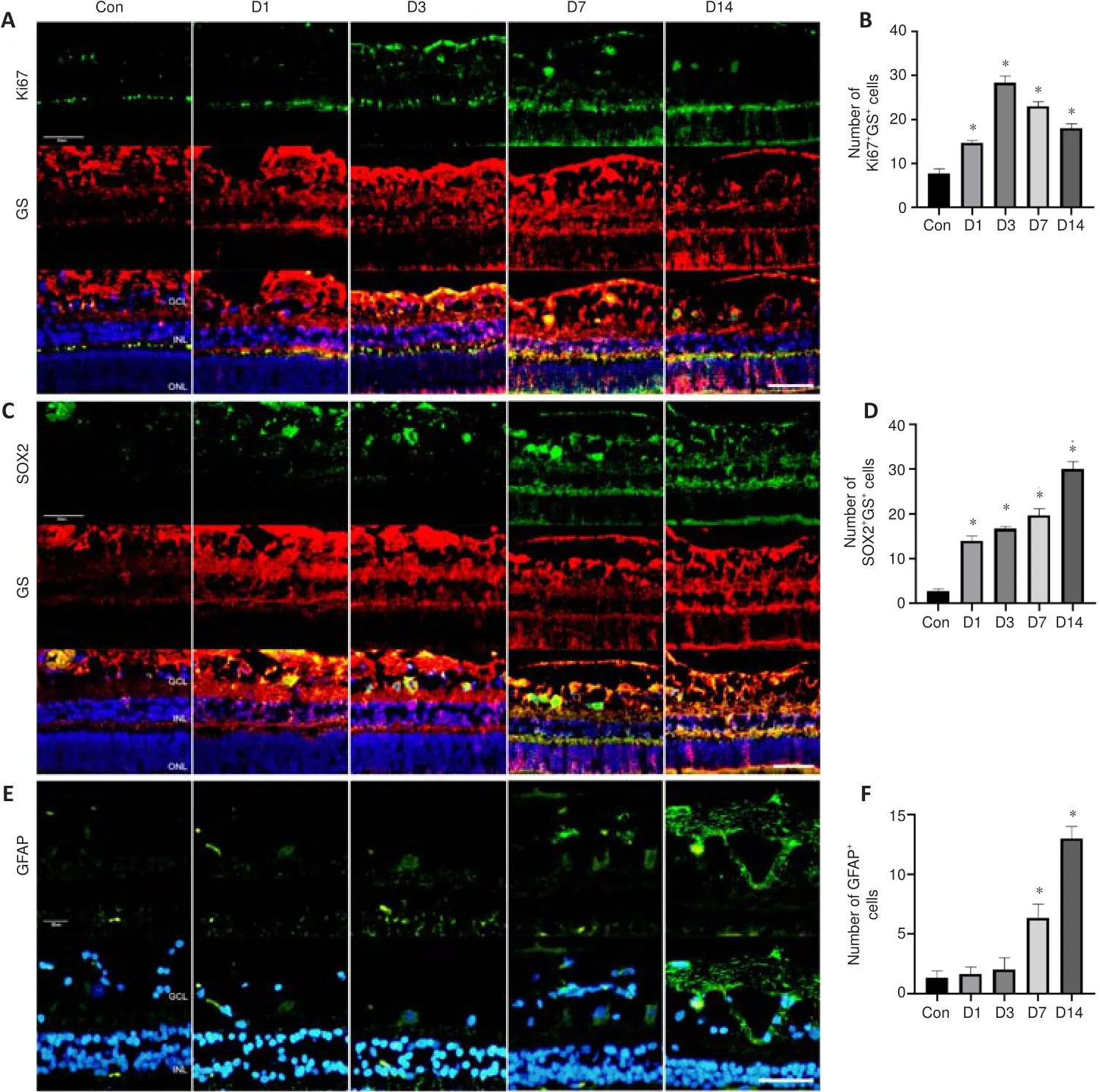
Figure 5|Müller cell regeneration and gliosis occur after blunt ocular trauma.
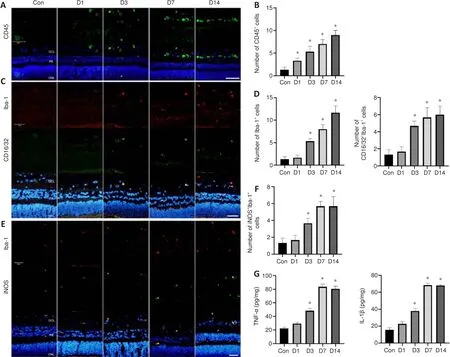
Figure 6|Inflammation and microglial polarization occur after blunt ocular trauma.

Figure 7 | ERG and F-VEP findings indicate impaired retinal function after blunt ocular trauma.
Discussion
In this study, we established a weight drop injury model of blunt ocular trauma in Beagle dogs.Only male Beagle dogs were used to avoid potential sex differences because such differences have been observed in human and mouse retinas (Bessinis et al., 2013; Trotta et al., 2021).Our results showed that the number of RGCs gradually decreased over time until 14 days after ocular trauma, while the thicknesses of the INL and ONL decreased until the seventh day after injury.Bipolar cells in the INL were most sensitive to blunt ocular trauma, which manifested as a decrease in the number of cells on the first day after injury.However, the numbers of other cells (e.g., cholinergic and amacrine cells) in the INL did not decrease until the seventh day;horizontal cells were not affected by blunt ocular trauma.Additionally, the cellular connections between bipolar cells and photoreceptor synapses were significantly reduced beginning on the seventh day after injury.Moreover,the extent of retinal cell necroptosis after injury increased over time; the necroptosis followed a pattern of progression from the GCL to the ONL.Visual electrophysiology findings indicated that visual impairment began on the first day after trauma and worsened over time.Finally, blunt ocular trauma induced nerve regeneration and Müller glial hyperplasia; it also resulted in the recruitment of microglia to the retina and polarization of those microglia to the M1 phenotype.
Pieramici et al.(1997) reported that patients with blunt ocular trauma may experience reduced vision.In that study, the authors presumed that vision problems were caused by retinal shock (e.g., Berlin edema) or more serious scleral/retinal injury (D’Orazi et al., 2014; Beier et al., 2017).However, the detailed temporal and spatial pathological changes of globe injury have not been fully characterized.In the present study, blunt ocular trauma caused the relative number of RGCs to decrease beginning on the first day after trauma, while the thicknesses of the INL and ONL decreased until the seventh day after injury.TUNEL staining showed that the progression of retinal cell death in ONL cells did not follow this pattern, indicating that damage in the GCL preceded damage in the INL and ONL.Moreover, we found that the number of RGCs significantly decreased beginning on the first day after injury,indicating that RGCs were more sensitive to external injury.RGCs constitute the end of the optic nerve and are present in the inner part of the retina;damage to these cells may be closely associated with vision loss after trauma.There are many types of cells in the inner part of the retina.Among them,bipolar cells serve as retinal interneurons that can receive signaling input from photoreceptors; after integration, they transfer these signal inputs to amacrine and ganglion cells (Beatty et al., 2000).In our study, we found that the number of bipolar cells significantly decreased beginning on the first day after trauma, whereas the numbers of other cells in the INL did not significantly decrease until the seventh day after injury.This difference may be related the strong plasticity of bipolar cells, which makes them more sensitive to cellular damage; such damage may be reversible because of the strong plasticity (D’Orazi et al., 2014; Beier et al., 2017).After damage to a subset of retinal photoreceptors, healthy photoreceptors might shift to the damaged area; adjacent bipolar cells could establish new connections with these healthy photoreceptors, thereby partially restoring retinal function (Blanch et al., 2012).As the most numerous interneurons, bipolar cells have more than a dozen different subtypes that may be connected to distinct visual pathways(Euler et al., 2014).Therefore, patients with ocular trauma can exhibit various manifestations of visual dysfunction.The retina could be divided into 10 different structures composed of various cells, which allows for different responses to trauma in various retinal areas.Our study found a pattern of retinal cell death after trauma, which progressed from the GCL to the ONL.Other studies have shown that post-trauma injury gradually diminishes from the center of the initial impact to the periphery of the impact in a horizontal manner (D’Orazi et al., 2014; Thomas et al., 2019), but the pattern of injury progression in the vertical direction has not been characterized.The characteristics of retinal cell death in the vertical direction might be influenced by ischemia, hypoxia, and the inflammatory response.In addition to the pattern of retinal cell death progression, the form of cell death has been controversial.Some studies demonstrated that the central area of the injury mainly exhibits necroptosis, while the peripheral areas of injury exhibit apoptosis (D’Orazi et al., 2014; Thomas et al., 2019).Notably, apoptosis only constitutes a fraction of the retinal injury that occurs after blunt ocular trauma(Bricker-Anthony and Rex, 2015), suggesting that necroptosis is the main form of cell death.In this study, the expression levels of necroptosis-related proteins greatly increased over time, which is consistent with the findings by Bricker-Anthony and Rex (2015) that the expression levels of necroptosisand apoptosis-related proteins increased over time after ocular blast injury, in combination with increased glial reactivity.However, in the study by Bricker-Anthony and Rex (2015), microglia were activated and expanded into the ONL at 3 days after injury and diminished at 7 days after injury; in our study,microglia were activated and continued to expand until the 14day after injury.These distinct microglial responses might be related to differences in trauma type.Bricker-Anthony and Rex (2015) used mild blunt trauma in their study.Animal models of blunt contusion injury were used in this study.Another study showed that reactive microglia remained in the ONL at 28 days after injury (Bricker-Anthony et al., 2014).Furthermore, Boehme et al.(2021)reported a decrease in RGC transcripts and an increase in glial cell transcripts at 7 days after injury, based on the results of RNA sequencing.Because there remains no clear model, the temporal and spatial changes in blunt ocular trauma manifest as distinct pathological states.
Because oxygen supply in the retina comes from two sources—retinal vasculature for the GCL and INL, and choroidal vasculature for the ONL—we used dark-adapted oscillatory potentials in F-VEP recordings to observe vascular conditions.Our results showed that after blunt ocular trauma, the total amplitude of oscillatory potentials decreased over time, indicating microcirculatory dysfunction that may result in retinal ischemic injury.Fu et al.(2021) reported the long-term results of ischemic injury in the rat eye,induced using a needle injection model.Their study mainly focused on high intraocular pressure and secondary retinopathy, as well as transneuronal degeneration and synapses.In our study, we used a weight drop injury model, similar to the air gun model used by Thomas et al.to induce blunt ocular trauma.However, our study focused on contusion, RGC death, and inflammatory response after retinal injury.Thus far, the literature has shown that the relative number of RGCs decreases after ocular ischemia or trauma.
Caspase-2 reportedly plays an important role in retinal injury caused by blunt ocular trauma (Thomas et al., 2018, 2021).The activation of caspase-2 might be associated with DNA damage and necrosis, a process that disrupts the normal cellular membrane structure, thus releasing cellular contents and activating the inflammatory response (Miles et al., 2017; Vigneswara and Ahmed, 2020); our findings are consistent with the previous reports.We also found that on the third day after trauma, increasing numbers of microglia were recruited to the retinal region; moreover, a large number of microglia were polarized to the M1 phenotype, along with enhanced releaseof inflammatory factors.Microglia were not activated on the first day after injury, suggesting that their activation is caused by necroptosis after trauma,rather than by ocular trauma.
There is evidence that necroptosis is associated with an inflammatory response (Ros et al., 2020).MLKL mediates cell membrane rupture during necroptosis, which leads to the release of cellular contents and the recruitment of macrophages that produce inflammatory factors (Xiong et al., 2016; Jin et al., 2017).M1 microglia, also known as pro-inflammatory macrophages, cause increased inflammation and exacerbate tissue damage.An extensive inflammatory response can cause damage to normal tissues(Xiong et al., 2016; Jin et al., 2017).Therefore, timely anti-inflammatory therapy might be necessary after trauma.Additionally, Müller cells in the retina are presumed to possess several characteristics of stem cells, which can proliferate and differentiate into neurons or glia after tissue injury.Our study showed that SOX2 transcription factor expression levels increased after injury,indicating that Müller cells were reprogrammed into stem cells and exhibited concomitant hyperplasia, suggesting that Müller cells have mixed effects after injury.While Müller cells can promote tissue repair, they can also promote the glial hyperplasia that impedes tissue regeneration (Rey-Funes et al., 2013; El-Azab et al., 2014; Sherpa et al., 2014).Therefore, further research is needed to determine how Müller cells might promote tissue regeneration.
Our findings suggest that necroptosis plays an important role in the exacerbation of retinal injury after blunt ocular trauma.However, the relationship between necroptosis and neuroinflammation has not been clearly elucidated.Subsequent experiments using chemical inhibitors or transgenic animals should be conducted to investigate whether interventions to manage necroptosis can affect the neuroinflammation caused by blunt ocular trauma, thereby alleviating retinal injury.In Beagle dog studies, it is difficult to perform genetic manipulation and chemical drug treatment due to a lack of pilot studies.Therefore, other animals such as mice and rats could be better animal models to conduct related studies.
In conclusion, this study showed that blunt ocular trauma can cause retinal injury and subsequent vision loss.Retinal cell death after trauma followed an inside-out pattern, which involved progression from the GCL to the ONL.Moreover, trauma caused retinal cell necroptosis, as well as the recruitment and polarization of microglia; these processes led to increased inflammation.The pathological changes in various retinal cells after blunt ocular trauma offer clues for future therapeutic strategies.
Author contributions:
FF and ZF designed and supervised this study and revised the manuscript.YH, XQW and TC conducted and performed the experiments, completed the analysis and wrote the initial manuscript.YND, BJ,XH and DYW performed the experiments.All authors have read and approved the final manuscript.
Conflicts of interest:
The authors declare that they have no competing interests.
Availability of data and materials:
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Open access statement:
This is an open access journal, and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewers:
Yongqing Liu, University of Louisville School of Medicine,USA; Steven Levy, MD Stem Cells, USA.
Additional files:
Results of power analysis.
Open peer review report 1.

- 中国神经再生研究(英文版)的其它文章
- Neural and Müller glial adaptation of the retina to photoreceptor degeneration
- Agomelatine: a potential novel approach for the treatment of memory disorder in neurodegenerative disease
- MicroRNAs: protective regulators for neuron growth and development
- In vivo astrocyte-to-neuron reprogramming for central nervous system regeneration: a narrative review
- Intranasal nerve growth factor for prevention and recovery of the outcomes of traumatic brain injury
- Altered O-GlcNAcylation and mitochondrial dysfunction,a molecular link between brain glucose dysregulation and sporadic Alzheimer’s disease