
Signaling interactions among neurons impact cell fitness and death in Alzheimer’s disease
2022-09-16 04:19CatherineYeatesPrajaktaDeshpandeMadhuriKangoSinghAmitSingh
中国神经再生研究(英文版) 2023年4期
Catherine Yeates , Prajakta Deshpande , Madhuri Kango-Singh , Amit Singh ,
Abstract The pathology of Alzheimer’s disease involves a long preclinical period, where the characteristic clinical symptoms of the changes in the brain are undetectable.During the preclinical period,homeostatic mechanisms may help prevent widespread cell death.Evidence has pointed towards selective cell death of diseased neurons playing a potentially protective role.As the disease progresses, dysregulation of signaling pathways that govern cell death contributes to neurodegeneration.Aberrant activation of the c-Jun N-terminal kinase pathway has been established in human and animal models of Alzheimer’s disease caused by amyloid-beta 42- or tau-mediated neurodegeneration.Clonal mosaic studies in Drosophila that examine amyloid-beta 42 in a subset of neurons suggest complex interplay between amyloid-beta 42-expressing and wild-type cells.This review examines the role of c-Jun N-terminal kinase signaling in the context of cell competition and short-range signaling interactions between amyloid-beta 42-expressing and wild-type neurons.Cell competition is a conserved phenomenon regulating tissue integrity by assessing the fitness of cells relative to their neighbors and eliminating suboptimal cells.Somatic clones of amyloid-beta 42 that juxtapose genetically distinct neuronal cell populations show promise for studying neurodegeneration.Generating genetic mosaics with labeled clones of amyloid-beta 42- or tau-expressing and wild-type neurons will allow us to understand how short-range signaling alterations trigger cell death in neurons and thereby contribute to the progression of Alzheimer’s disease.These approaches have the potential to uncover biomarkers for early Alzheimer’s disease detection and new therapeutic targets for intervention.
Key Words: Alzheimer’s disease; amyloid-beta 42 mediated neurodegeneration; cell competition;Drosophila; c-Jun N-terminal kinase signaling; suboptimal cell; super competition; super competitor cell; two clone-approach; wild type cell
Introduction
Alzheimer’s disease (AD) is a progressive neurodegenerative disease predominantly affecting people older than 65 (Knopman et al., 2021).During the onset of disease, impairments in memory and cognitive function occur,ultimately resulting in the death of the individual (McKhann et al., 1984;Knopman et al., 2021).Widespread cell death leads to a shrinkage in the size of the cortex, concomitant with cognitive function impairments seen in the disease (McKhann et al., 1984; Shankar et al., 2008; Knopman et al., 2021).Yet, there is a long latency period before clinical symptoms appear, and the changes to the brain occurring prior to symptom manifestation have not been well understood (Hardy, 2009; Yeates et al., 2019).To date, there is no cure for AD, and only a few effective treatments have been developed.The vast majority of pharmaceutical treatments fail along the path from initial drug development and testing to animal models and finally, clinical trials (Goldman et al., 2018; Deshpande et al., 2019).
The mechanisms underlying disease etiology and progression have been a subject of considerable study.The hallmarks of AD are extracellular accumulation of amyloid-beta 42 (Aβ) plaques and intracellular accumulation of tau protein in neurofibrillary tangles.Aβ, a 42 amino acid polypeptide,is generated after amyloid precursor protein is cleaved by β-secretase and γ-secretase (Glenner and Wong, 1984; Masters et al., 1985; Siegel et al.,2017).Familial AD is associated with mutations in amyloid precursor protein and the presenilin 1 and presenilin 2 genes (Magee, 1995; Lleó et al., 2002;Eggert et al., 2004; Kelleher and Shen, 2017).Aβis hydrophobic in nature and prone to aggregation, forming extracellular Aβplaques (Hickman et al., 2016; Sarkar et al., 2016).Cell death, a hallmark of neurodegenerative disorders (Singh, 2012), occurs following the accumulation and aggregation of Aβplaques (Table 1; Tare et al., 2011; Sarkar et al., 2016; Cline et al., 2018;Yeates et al., 2019, 2020).Deposition of Aβplaques follows a sequence,affecting the neocortex first, followed by the hippocampus and other brain regions (Thal et al., 2002; Sengoku, 2020).However, disease progression is complex as further cell signaling changes occur downstream of Aβand neurofibrillary tangle accumulation (Cline et al., 2018).

Table 1 |Glossary
A substantial body of research suggests that AD’s progression involves aberrant activation of signaling pathways that are normally tightly controlled, including those that regulate cell death.In this review, we will focus on alterations to signaling pathways that occur downstream of Aβaccumulation.Similar approaches are being pursued downstream of the expression of hyperphosphorylated forms of tau inDrosophila
(Singh et al.,unpublished data).The evolutionarily conserved c-Jun N-terminal Kinase (JNK)pathway is one such pathway implicated in animal models of AD (Tare et al.,2011; Yeates et al., 2019; Irwin et al., 2020).JNK signaling plays an important role in development.Aberrant activation of JNK occurs during stressresponse, cell competition, and neurodegeneration, ultimately leading to cell death (Sun et al., 2022).An increase in JNK phosphorylation and activation has been reported in AD patients (Wang et al., 2014; Yarza et al., 2015), and robust evidence from animal models points to JNK activation as a key part of the neurodegeneration observed downstream of Aβaccumulation (Tare et al., 2011; Yeates et al., 2020; Gogia et al., 2021).However, studies have largely focused on models in which Aβis expressed uniformly throughout neuronal tissue (Cao et al., 2008; Tare et al., 2011; Moran et al., 2013;Sarkar et al., 2018; Yeates et al., 2019; Irwin et al., 2020).As a result, there is no information on how Aβ-expressing versus wild-type neuronal cell populations respond to Aβaccumulation.Given the multitude of signaling pathways implicated in AD pathology and the progressive nature of the disease, it is necessary to understand how signaling dynamics between cell populations contribute to disease pathology (Yeates et al., 2019, 2020).Crosstalk among cells and their neighbors occurs through secreted signals,activation of intracellular signaling pathways as well as expression of cell surface markers.Cell competition is one such process implicated both in AD and in cell death mediated by JNK signaling.Cell competition is a process where the juxtaposition of genetically different cells (which are individually viable and contribute to tissue or organ development) causes the elimination of the less fit group of cells by active signaling from the more fit neighbors (Table 1)(Morata and Ripoll, 1975; Baker, 2020).Differential expression of cell surface markers and signaling pathway activity determines cellular fitness, and explains how less fit cells are eliminated.Determination of cell fitness depends on the context, with certain mutations resulting in super-competitor cells that proliferate while wild-type neighbors are eliminated (Table 1; Moreno and Basler, 2004).Introducing cell competition into our conceptualization of AD may help us understand the role of cell population dynamics in neurodegeneration.Neurodegeneration in AD begins on a smaller scale, affecting fewer areas of the brain, and progressively affects more brain regions.By investigating local signaling changes and cell death in AD models, we gain a more comprehensive picture of disease progression and find new targets for intervention.
Search Strategy and Selection Criteria
We searched PubMed for studies published from 2012 to 2022 using the following keywords: Alzheimer’s disease, neurodegeneration, neuronal cell death, cell competition, super competition, FLP/FRT,Drosophila
, MARCM,mosaic analysis, JNK signaling, and two clone-approach.Drosophila Melanogaster as a Model for Alzheimer’s Disease
The fruit fly,Drosophila melanogaster
, is an excellent model to study neurodegenerative diseases (Pandey and Nichols, 2011; Singh and Irvine,2012; Fernandez-Funez et al., 2013; Bolus et al., 2020).Approximately 70%of the genes associated with human disease have homologs inDrosophila
(Reiter et al., 2001; Bier, 2005), and many components of synapse structure and neurotransmission are conserved between flies and humans (McGurk et al., 2015; Sarkar et al., 2016; Yeates et al., 2019).Furthermore, flies are wellsuited to genetic and pharmacological screens, with numerous large-scale screens yielding new insights into genetic modifiers (Pandey and Nichols,2011; Moran et al., 2013; Steffensmeier et al., 2013) and new candidate drugs for AD (Fernandez-Funez et al., 2013; Deshpande et al., 2019).Disease models inDrosophila
exploit the power of its genetics, its amenability to a variety of mutagenesis techniques, and its ability to express foreign genes to mimic several important neurodegenerative disorders in the compound eye(Fortini et al., 2000; Bonini and Fortini, 2003; Hirth, 2010; Bolus et al., 2020).In addition,Drosophila
is amenable to chemical screens - thus allowing a quick and relatively inexpensive whole-animal model for testing inhibitors of the AD neurodegeneration phenotype (Gladstone and Su, 2011; Gonsalves et al., 2011; Tello et al., 2022).Advantages of Modeling Alzheimer’s Disease in the Drosophila Eye
Drosophila
eye serves as an ideal organ system to assay the effects of neurodegeneration as (a) the genes involved in eye development exhibit structural and functional similarities between insects and humans, (b) eyes are not essential for the viability or fertility of the fly (Fortini et al., 2000;Pandey and Nichols, 2011; Rincon-Limas et al., 2012; Singh and Irvine, 2012;Singh et al., 2012; Tare et al., 2013; Gogia et al., 2020), and (c)Drosophila
has a fully functional nervous system with an architecture that separates specialized functions such as vision, olfaction, learning, and memory.This allows functional and behavioral studies (e.g., investigating neurotransmission or memory) inDrosophila
neurodegeneration models.The adult compound eye ofDrosophila
consists of 800-unit eyes and develops from the eye imaginal disc of the larva (Figure 1; Garcia-Bellido and Merriam,1969; Haynie and Bryant, 1986; Tare et al., 2013), which is a very well-studied system in terms of patterning, growth regulation, neural development and modeling human disease (Ready et al., 1976; Singh and Irvine, 2012; Singh et al., 2012; Tare et al., 2013; Gogia et al., 2020).Retinal precursor cells are differentiated into photoreceptor clusters and restricted to the region posterior to the furrow (Wolff and Ready, 1993; Kumar, 2011).Thus, theDrosophila
eye serves as an excellent model to study cell death in AD (Tare et al., 2011; Figures 1 and 2).The targeted misexpression genetic strategy of the Gal4-UAS (upstream activation sequence) system (Brand and Perrimon, 1993) is commonly used to model human diseases inDrosophila
.This system allows the expression of a transgene (e.g., human Aβ) in a specific tissue of interest (e.g.,Drosophila
retina).Transgenic expression of Aβin flies recapitulates many aspects of disease pathology including amyloid plaque formation and aggregation,cell death, and defects in learning and memory (Iijima et al., 2004; Cao et al., 2008; Hirth, 2010; Tare et al., 2011; Moran et al., 2013; Steffensmeier et al., 2013; Sarkar et al., 2018; Irwin et al., 2020).One of the models of AD involves driving Aβexpression in the developing retina using the enhancer of glass multiple repeat (GMR).Here, Gal4 binds UAS conjugated to human Aβ, triggering its expression in retinal neurons (Cao et al., 2008;Tare et al., 2011; Singh and Irvine, 2012; Yeates et al., 2019).Misexpression of Aβin developing retinal neurons of the fly eye results in progressive neurodegeneration (Tare et al., 2011; Figure 2).Other AD models utilize different Gal4 drivers or express tau or other genetic lesions linked to this disease inDrosophila
.However, among these, the Aβmodel is a wellestablished model and a key focus of this review.Aberrant c-Jun N-Terminal Kinase Signaling in Alzheimer’s Disease
Aβ42 accumulation triggers neurodegeneration through aberrant activation of signaling pathways.Previous studies suggest upregulation of JNK signaling triggers neuronal cell death in AD (Tare et al., 2011; Yarza et al., 2015; Sarkar et al., 2018; Irwin et al., 2020).JNK signaling is a conserved pathway betweenDrosophila
and mammals (Geuking et al., 2005) and is activated in apoptosis,cell growth, inflammation, and immune response.JNK signaling is activated by binding of the ligand Eiger (Egr) to its receptor(s) Wengen (Wgn) and/or Grindelwald (Grnd) (Kanda et al., 2002; Moreno et al., 2002).Activation of the receptors induces a cascade of kinases likehemipterous
(hep
) theDrosophila
JNKK andbasket
(bsk
), theDrosophila
JNK (Glise et al., 1995;Sluss et al., 1996; Holland et al., 1997).Bsk phosphorylates and activates the transcription factor Jun that is then translocated to the nucleus and induces the expression of JNK target genes (Sluss et al., 1996; Kockel et al., 2001).Puckered (Puc), a dual-specificity phosphatase, is the transcriptional target of JNK signaling and negatively regulates the JNK signaling pathway (Martín-Blanco et al., 1998; Stronach, 2005).JNK signaling induces cell death both through caspase-dependent mechanisms by activating pro-apoptotic factors likehead involution defective
(hid
),reaper
(rpr
), grim, and through caspaseindependent mechanisms (Martín-Blanco et al., 1998; Singh et al., 2006).JNK phosphorylates tau and amyloid precursor proteinin vitro
(Yoon et al., 2012; Sun et al., 2022), thereby promoting the accumulation of hyperphosphorylated tau and Aβ.InDrosophila
, misexpression of Aβin neurons of the brain leads to impaired locomotor function, age-dependent learning defects, progressive loss of neurons, and reduced lifespan (Iijima et al., 2004; Cao et al., 2008; Hirth, 2010; Tare et al., 2011; Moran et al., 2013;Steffensmeier et al., 2013; Cutler et al., 2015; Irwin et al., 2020).Earlier,it has been reported that Aβinduces aberrant cellular morphology and increased cell death in the developing retinal neurons in the late third instar eye imaginal disc.JNK signaling is aberrantly activated in neurons where Aβis misexpressed, implicating its role in Aβ-mediated neurodegeneration(Tare et al., 2011; Irwin et al., 2020).Activation of JNK signaling exacerbated Aβneurotoxicity, whereas downregulation of the JNK pathway prevented cell death and rescued eye size (Tare et al., 2011).In addition to triggering neurodegeneration, JNK is notable for its physiological role in the process of cell competition, a field of study that is garnering more interest for its potential role in AD (Kucinski et al., 2017).Do Cell Competition and/or Intercellular Interactions Drive Neurodegeneration?
Cell competition is involved in the regulation of tissue integrity and homeostasis and has important implications in several diseases linked to impaired proliferation and/or survival, including neurodegeneration and cancer (Figure 3 and Table 1; Coelho and Moreno, 2019; Marques-Reis and Moreno, 2021; Morata, 2021; Maruyama and Fujita, 2022).Although the phenomenon of cell competition was first described by Morata and Ripoll while studying the interactions between Minute and normal cells (Morata and Ripoll, 1975), recent studies on the molecular mechanisms underlying cell competition revealed the role of membrane proteins.Differential expression of membrane proteins mediates information on cell fitness status,promoting apoptosis when cells are judged to be less fit than their neighbors.On the other hand, in tumorigenesis studies, cells with higher levels of Myc become super-competitors, leading to the elimination of their wild-type(WT) neighbors (Figure 3; Moreno and Basler, 2004; Parker et al., 2021).Interestingly, it has been suggested that cell competition may play a role in AD (Coelho et al., 2018; Coelho and Moreno, 2019; Yeates et al., 2020).The connections between cell competition, AD, and JNK-mediated cell death suggest that a complete understanding of interactions between neighboring cells may shed light on disease progression in AD.
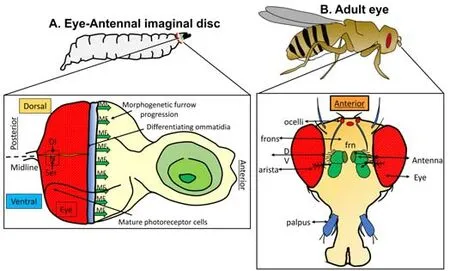
Figure 1|Drosophila eye model to study patterning and disease.

Figure 2 | Drosophila eye as a model for Alzheimer’s disease.
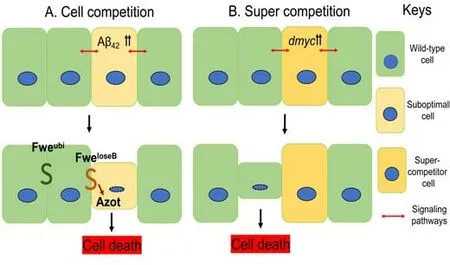
Figure 3 | Schematic representation of cell competition and super competition.
Cell competition was first described inDrosophila melanogaster
.The original study examinedMinute
mutants, which have impaired ribosomal activity due to mutations in ribosomal protein genes (Morata and Ripoll, 1975; Marygold et al., 2007; Akai et al., 2018; Baumgartner et al., 2021).HeterozygousMinute
flies have a short developmental delay.In genetic mosaics of wild-type and heterozygousMinute
cells, theMinute
cells were eliminated (Morata and Ripoll, 1975).A similar type of cell competition has been identified in bothDrosophila
and mammals in homozygousMahjong
mutants (Tamori et al.,2010).In theDrosophila
wing disc,Mahjong
-knockout cells are eliminated when surrounded by wild-type cells.Mahjong
knockdown cells in mammalian cell culture are similarly eliminated.In both flies and mammals, elimination of these cells occurs through JNK-mediated apoptosis (Tamori et al., 2010; Kajita and Fujita, 2015).The transmembrane protein Flower (Fwe) is a key marker of cell fitness,determined through differential expression of three splice isoforms,Fwe, Fwe, and Fwe(Coelho and Moreno, 2019).Under normal circumstances, Fweis ubiquitously expressed.Downregulation of Fweassociated with low fitness leads to upregulation of the Fweisoforms.Cells are marked for death and eliminated when they express the Fweisoform relative to neighboring cells (Rhiner et al., 2010).In addition to Fwe,theDrosophila
homolog of Secreted Protein, Acidic, Cysteine -Rich introduces an additional layer of regulation of cell competition.When transcriptionally upregulated in suboptimal “loser” cells,dsparc
inhibited caspase activation,potentially allowing cells to recover from a transient reduction in their fitness(Portela et al., 2010).Downstream of Fwe and Secreted Protein, Acidic,Cysteine-Rich, transcription ofahuizotl
(azot
) serves as a fitness checkpoint prior to initiation of pro-apoptotic signaling.Furthermore,azot
knockouts showed reduced lifespan, providing further evidence that cell competition can promote tissue health by eliminating less fit cells (Merino et al., 2015).Elimination of cells by cell competition and/or intercellular interactions has important implications for understanding the progression from the long asymptomatic phase of AD to the presentation of clinical symptoms.Deposition of Aβcan be detected over many years prior to the onset of clinical symptoms in AD (Mufson et al., 2016).Moreover, higher levels of amyloid deposition in clinically normal subjects correlate with lower scores on episodic memory tests (Sperling et al., 2013).A wealth of evidence points to complex alterations to cells and cellular networks in AD; however, compensatory mechanisms may mask these alterations,delaying the onset of clinical symptoms (De Strooper and Karran, 2016).The elimination of suboptimal neurons early in AD could delay the onset of widespread neurodegeneration and hence may play a protective role against AD.Enhancing this selective cell death before neurodegeneration becomes widespread, could be beneficial in treating AD (Coelho et al., 2018;Coelho and Moreno, 2019).In addition to the role cell competition and intercellular interactions may play in the asymptomatic phase of AD, insights from these studies may help explain the molecular underpinnings of AD neurodegeneration.
Context-Dependent Roles of Cell Competition and Intercellular Interactions
Cell competition is highly context-dependent and can lead to the death of either wild-type or mutant cells under different circumstances.Preferential death of wild-type cells was observed in our recent study on neurodegeneration in AD (Yeates et al., 2020).In cell competition studies, the death of wild-type cells is observed in cancer models.Further, cell competition can either contribute to or defend against mechanisms involved in cancer(Kanda and Igaki, 2020).Loss of the cell polarity genescribble
(scrib
) results in overproliferation of mutant tissue.Clones ofscrib
mutant tissue surrounded by wild-type cells fail to proliferate; the wild-type cells prevent proliferation by increasing JNK-mediated apoptosis of mutant tissue.However,scrib
mutants did proliferate when oncogenic mutations in Ras or Notch were added(Brumby and Richardson, 2003).Thus, oncogenic cooperation showed a role in cell competition in tumor suppression.Alternatively, super-competition is a phenomenon associated with tumor growth where oncogenic cells proliferate while wild-type cells are eliminated.Cells expressing higher levels of the proto-oncogenedMyc
become super-competitors, with neighboring wild-type cells judged as less fit in comparison (Figure 3; Moreno and Basler, 2004).Similar super-competition has also been described for Hippo pathway mutants that lead to activation of Yorkie, with evidence supporting a bistable positive feedback loop between Yorkie and Myc (Figure 3; Neto-Silva et al., 2010;Ziosi et al., 2010).Thus, cell competition may preserve or eliminate wild-typetissue depending on the cellular context.Recent research has delved into the links between cell competition and neurodegeneration and implicated JNK signaling in the death of mutant cells and wild-type cells depending on the cellular context.Models for studying short-range signaling interactions such as the process of cell competition may yield new insights into the progression of AD.New Approaches to Understand Aberrant Cell-Cell Interactions in Alzheimer’s Disease
Recently, we expanded upon existingDrosophila
models of AD by creating a “two-clone” system to study the signaling interactions between Aβ42-expressing and WT neurons (Figure 4; Yeates et al., 2020).This system uses mitotic recombination by combining flippase/flippase recognition target (FLP/FRT) and the Gal4/UAS/Gal80 system to generate mosaics in developing retinal neurons (Figure 4; Xu et al., 1995; Blair, 2003).A cross producesDrosophila
larvae with the following genetic components: a Flippase expressed under aheat shock promoter (hs-FLP), one copy of green fluorescent protein (GFP)under a ubiquitin promoter (ubi-GFP), one copy of Gal80 under a tubulin promoter (TubGal80), and Aβexpressed under the control of the GMRGal4 driver (GMR>Aβ).Therefore, GMR>Aβdrives the expression of Aβin neurons of the developing retina.Gal80 is a repressor protein that binds Gal4 and renders it inactive (Yeates et al., 2020).These components used in combination with the FLP/FRT system lead to the generation of two labeled cell populations.The Flippase directs recombination between FRT sites, here located adjacent to ubi-GFP and TubGal80.The cross generates progeny with the full genotype hsflp; GMR>Aβ42/+; FRT82BTubGal80/FRT82Bubi-GFP.A heat shock at 37°C triggers mitotic recombination at the FRT sites, which results in two clonal populations of neurons: one cell population that expresses the GFP reporter and Aβin developing retinal neurons and the other cell population is WT.Both the Aβ-expressing clone (GFP positive) and WT sister clone (GFP negative) are easily distinguished from the background, which weakly expresses GFP, facilitating quantification of the clone size (Figure 4).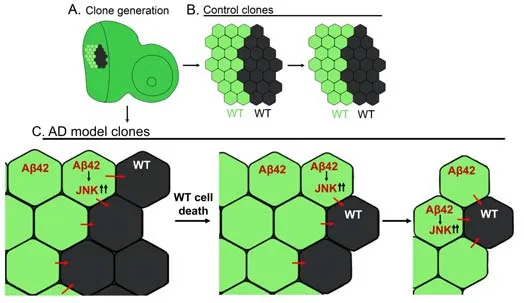
Figure 4| A two-clone system to study interactions between Aβ42-expressing and WT cell populations.
Because these two clones originate from a single progenitor cell, one would expect them to be equivalent in size.Indeed, in the absence of Aβ, sister clones were not significantly different in size (Figure 4).However, in the presence of Aβexpressing clones, it was wild-type clones that were smaller suggesting a more complex interplay between Aβ-expressing and WT cells(Figure 4).Such differences in clone size may be related to differential levels of cell death.A comparison of cell death revealed that many more cells were dying in the WT clones.Thus, Aβ-expressing clones and their WT sister clones start under similar conditions, but WT cells die first, followed by Aβ-expressing cells (Yeates et al., 2020; Figure 4).
The JNK signaling pathway is implicated in cell death observed in fly eye model and in mammalian AD models (Jeon et al., 2020).The two-clone system demonstrated an activation of JNK signaling in Aβ-expressing clones in comparison to WT clones.Furthermore, misexpression of a constitutively active form of the JNK kinase homolog in flies,hemipterous
(hep
) in Aβ-expressing clones (Glise et al., 1995; Tournier et al., 1997)resulted in significantly smaller WT clones in comparison to their sister clones expressing both Aβandhep
.On the contrary, decreasing JNK activity in Aβ-expressing clones by misexpression of a dominant-negative form ofDrosophila
JNK,basket
(bsk
) in Aβ-expressing clones restored the size of WT sister clones.These studies provided evidence for complex crosstalk between WT and Aβ-expressing clones, ultimately leading to cell death mediated at least in part by the JNK pathway.Though cell death occurs in Aβ-expressing clones, it appears to occur later.Interactions between these clones lead to sensitization of WT cells and eventually cell death, resulting in smaller WT clones (Figure 4).The study of cell competition in AD has uncovered evidence for selective cell death of Aβ-expressing neurons (Coelho et al., 2018; Coelho and Moreno,2020; Costa-Rodrigues et al., 2021).Upregulation of markers of loser cell status, Fwe, and Azot, was reported following uniform expression of Aβthroughout theDrosophila
retina.Then Aβwas expressed in small clones within the neuroepithelium of the fly eye disc.Flowerand Azot were upregulated within these clones, indicative of lower fitness, and the clones were eliminated over time by apoptosis (Coelho et al., 2018; Figure 3).However, clones in this system were small, and Aβ-expressing and WT clones were not labeled and examined within the same eye disc.Fweand cell death marker DCP-1 were observed outside of the Aβ-expressing clones as well.On the whole, this selective death of Aβ-expressing cells had a protective effect (Coelho et al., 2018).This was seen in related research on neuronal circuits, in which selectively removing suboptimal cells increases the function of the circuit as a whole: hyperactive neurons may be eliminated through cell competition, restoring circuit function (Coelho and Moreno,2020; Costa-Rodrigues et al., 2021).For cell competition to protect against neurodegeneration in AD, diseased neurons must be correctly identified as less fit and eliminated via an early compensatory mechanism before pathological changes accumulate.
Conclusion
AD is a multifactorial disease with no cure (Yeates et al., 2019; Jeon et al.,2020).Since neurons are post-mitotic cells and adult neurogenesis is limited,early detection of neurodegeneration is paramount to preserving as much cognitive function as possible.Understanding the molecular mechanisms underlying AD will give us better tools to delay or prevent widespread neurodegeneration.
Substantial evidence shows that dysregulation of physiological signaling pathways contributes to the etiology and progression of AD.JNK signaling plays important roles in development, including nervous system development and neuronal pruning (Bornstein et al., 2015; Schellino et al., 2019; Zhu et al., 2019).Aberrant activation of JNK signaling is involved in the widespread neurodegeneration seen in AD.In the two-clone system, JNK signaling is upregulated in Aβ-expressing clones concomitant with the elimination of wild-type cells; blocking JNK in Aβ-expressing clones prevented the death of WT clones (Yeates et al., 2020).These findings suggest the utility of components of the JNK signaling pathway as biomarkers.
The selective cell death that occurs during cell competition can improve tissue health overall.However, when JNK signaling and selective cell death are leftunchecked, they contribute to the neurodegeneration seen in AD (Wang et al., 2014; Yarza et al., 2015).Research in cancer models demonstrates that cell competition can result in the death of wild-type cells, facilitating the proliferation of cancer cells.Cell competition and other processes regulating tissue homeostasis could protect against damage from Aβplaques accumulation early on in AD only to effectively contribute to cell death and widespread neurodegeneration later in disease progression.
Clone models have promising utility to study the short-range signaling interactions contributing to neurodegeneration.Understanding the interplay between JNK signaling and cell competition may allow us to determine which molecules could best function as biomarkers in AD (Sharma et al., 2021).Cell fitness marker Fwemay have utility as a biomarker of early-stage AD and requires further investigation.Azot is a fitness sensor, which induces expression of pro-apoptotic factorhead involution defective
(hid
) (Costa-Rodrigues et al., 2021).These components may also serve as biomarkers or therapeutic targets.Furthermore, emerging research has linked changes in microRNA expression with both AD and JNK-mediated neurodegeneration,generating interest in microRNAs as biomarkers (Angelucci et al., 2019; He et al., 2020).Selective cell death may help protect against neurodegeneration in AD, and for this reason, the molecular players underlying cell competition should be considered potential therapeutic targets.Understanding the conditions which prime cells for selective cell death or progressive neurodegeneration will improve our range of therapeutic targets and biomarkers for AD.Acknowledgments:
We thank Bloomington Drosophila Stock Center (BDSC)for the Drosophila strains, Developmental Studies Hybridoma Bank (DSHB)for the antibodies and members of Singh lab for providing comments on the manuscript.Confocal Microscopy was supported by the core facility at University of Dayton.
Author contributions:
CY, PD, MKS, and AS were involved in developing the concept, design, writing and editing of the manuscript.All authors approved the final version of the manuscript.
Conflicts of interest:
The authors declare no conflicts of interest.Editor note: AS is an Editorial Board member of Neural Regeneration Research.He was blinded from reviewing or making decisions on the manuscript.The article was subject to the journal’s standard procedures, with peer review handled independently of this Editorial Board member and their research groups.
Availability of data and materials:
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Open access statement:
This is an open access journal, and articles are distributed under the terms of the Creative Commons
AttributionNonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the
identical terms.
Open peer reviewer:
Ashu Johri, Weill Cornell Medical College: Weill Cornell Medicine, USA.
Additional file:
Open peer review report 1.

- 中国神经再生研究(英文版)的其它文章
- Neural and Müller glial adaptation of the retina to photoreceptor degeneration
- Agomelatine: a potential novel approach for the treatment of memory disorder in neurodegenerative disease
- MicroRNAs: protective regulators for neuron growth and development
- In vivo astrocyte-to-neuron reprogramming for central nervous system regeneration: a narrative review
- Intranasal nerve growth factor for prevention and recovery of the outcomes of traumatic brain injury
- Altered O-GlcNAcylation and mitochondrial dysfunction,a molecular link between brain glucose dysregulation and sporadic Alzheimer’s disease