
高效液相色谱-三重四极杆质谱法测定土壤和沉积物中六溴环十二烷和四溴双酚A
2022-09-16 07:08:06朱超飞杨文龙殷也筑范红利吕美玲
环境科学研究 2022年9期
朱超飞,杨文龙,殷也筑,杨 茜,杜 兵*,范红利,吕美玲
1. 生态环境部环境发展中心,国家环境分析测试中心,国家环境保护二英污染控制重点实验室,北京 100029
2. 安捷伦科技(中国)有限公司,北京 100102
六溴环十二烷(HBCDs)和四溴双酚A (TBBPA)是两类用量最大的溴代阻燃剂(BFRs),前者广泛用于建筑材料、车辆和纺织品中膨胀聚苯乙烯(EPS)和挤压聚苯乙烯(XPS)的阻燃,也包括室内装饰纺织品、家用电器、塑料和其他材料的阻燃,后者不仅可以作为反应型阻燃剂制造溴化环氧树脂、酚醛树脂和含溴聚碳酸酯,也作为添加型阻燃剂用于丙烯腈-丁二烯-苯乙烯(ABS)和高抗冲聚苯乙烯(HIPS)树脂中. HBCDs 理论上存在16 种立体异构体,产品中主要由α-HBCD、β-HBCD 和γ-HBCD 构成,三者含量分别为10%~13%、1%~12%和75%~89%.TBBPA 结构中含有2 个酚羟基,具有疏水性,其p和p分别为7.5 和8.5. HBCDs 和TBBPA 在生产、使用、处理和回收过程中均会泄露进入环境. 生产企业周边以及南北极地区的土壤和沉积物中均检出HBCDs 和TBBPA,浓度范围在未检出至几千μg/kg 之间. HBCDs 和TBBPA 具有持久性、生物富集性、长距离迁移性和毒性等特征. 2013 年,HBCDs 被列入《关于持久性有机污染物的斯德哥尔摩公约》附件A,2016 年《〈关于持久性有机污染物的斯德哥尔摩公约〉新增列六溴环十二烷修正案》正式在我国生效,截至2021 年底,我国已经全面禁止HBCDs 的生产、使用和进出口. 尽管TBBPA 在我国尚未得到管控,但作为仅次于HBCDs 的阻燃剂,存在类似的环境风险,需要开展广泛调查.
我国HBCDs 和TBBPA 监测技术体系研究起步较晚,缺乏针对土壤和沉积物中HBCDs 和TBBPA的监测标准,制约了区域性和系统性环境监测工作的推进. 随着“十四五”期间有关“加强新污染物治理”工作的逐步深入,加快推进HBCDs 和TBBPA 的标准化工作迫在眉睫.
研究显示,我国发布的涉及HBCDs 和TBBPA 的标准分析方法主要集中在食品、塑料和电子电气领域,除了海洋行业标准HY/T 260-2018《海洋沉积物体中六溴环十二烷的测定 高效液相色谱-串联质谱法》涉及海洋沉积物中HBCDs 的测定之外,主要国家、地区及国际组织尚未发布有关土壤和沉积物中两类目标物同时测定的标准分析方法.目前,已有较多文献涉及样品采集至分析测试的全流程. 样品的提取方式包括索氏提取、加压流体提取、超声振荡提取、微波辅助萃取等,常用的溶剂为丙酮与正己烷(体积比为1∶1)、正己烷与二氯甲烷(体积比为1∶1)等. 净化方式主要有酸性、中性和碱性的复合硅胶填充柱,单一中性硅胶填充柱,中性与酸性硅胶结合的填充柱,以及硅胶、C和HLB 等固相萃取小柱. 仪器分析方法以液相色谱-串联质谱为主,该方法分离检测过程中的温度可以控制在160 ℃以下,避免HBCDs 异构体之间的转换,从而实现异构体的特异性分析,且对于同时测定的TBBPA,也不需要如气相色谱测试前的衍生化操作. 研究表明,这两类物质的定量方法以内标法和同位素稀释法为主.
目前,研究多集中在对单一介质或一类目标物的分析,且前处理方法和仪器条件存在差别. 该研究在借鉴已有研究的基础上,综合评价目标物在土壤,尤其是基质复杂的沉积物的提取方式,建立了同步测定土壤和沉积物中HBCDs 和TBBPA 的高效液相色谱-三重四极杆质谱分析方法,以期服务于履约和新污染物监测的需求.
1 材料与方法
1.1 试剂与耗材
色谱纯有机溶剂包括丙酮、正己烷、二氯甲烷、乙腈和甲醇购自霍尼韦尔(Honeywell, 新泽西州, 美国). 4 种 目 标 物(α-HBCD、β-HBCD、γ-HBCD 和TBBPA)、同位素替代物(C-α-HBCD、C-β-HBCD、C-γ-HBCD 和C-TBBPA)、进样内标(D-α-HBCD)购自剑桥同位素实验室(Cambridge Isotope Laboratories,美国). 硅胶、优级纯乙酸铵(纯度>98%)、氨水和甲酸购自Acros Organics (Belgium, 美国). 优级纯硫酸钠购自国药集团化学试剂有限公司. 纯水由实验室自制(18 MΩ·cm). 44%酸性硅胶由78.6 g 浓硫酸(98%)和100 g 中性硅胶混合得到,2% KOH 碱性硅胶由40 mL 50 g/L 的KOH 水溶液与100 g 硅胶混合得到. 复合硅胶柱从下至上填充方式依次为1 g 无水硫酸钠、1 g 中性硅胶、10 g 的44%酸性硅胶和1 g 无水硫酸钠. 玻璃填充柱的柱长260 mm,内径15 mm.
1.2 样品前处理
土壤和沉积物样品经冷冻干燥,研磨粉碎后过60 目(0.25 mm)筛网,称量10.00 g,加入同位素替代物内标后进行加压流体提取,溶剂为丙酮与正己烷(体积比为1∶1),加热温度为100 ℃,静态萃取5 min,萃取压力设定为1.03×10Pa,循环萃取3 次. 提取液由复合硅胶柱净化,用100 mL 正己烷与二氯甲烷(体积比为1∶1)洗脱,洗脱液氮吹转溶至1 mL 甲醇,添加进样内标,过0.22 μm 滤膜后,上机分析.
1.3 仪器分析方法
该研究使用高效液相色谱-三重四极杆质谱(1290 Infinity Ⅱ LC-6470 MS/M S 系 统,Agilent, 美国)测定样品中3 种HBCDs (α-HBCD、β-HBCD 和γ-HBCD)和TBBPA 的浓度. 液相色谱柱为Zorbax RRHD Eclipse Plus C(2.1 mm×100 mm, 1.8 μm,Agilent, 美国),流动相为纯水和乙腈,流速为0.30 mL/min,柱温为35 ℃,进样量为5.0 μL. 基于电喷雾负离子化(ESI)和多反应监测(MRM)模式对目标物进行检测. HBCDs 监测离子对均为[M-H]->Br模式,自然态的监测离子对为640.8->81.0 和640.8->79.0,同位素替代物的为652.8->81.0 和652.8->79.0,进样内标D-α-HBCD 的 监 测 离 子 对 为657.8->81.0 和657.8->79.0;TBBPA 及C-TBBPA 的监测离子对分别为542.7->417.9 和542.7->291.0 及554.7->430.7 和554.7->296.7. 采用同位素稀释法的平均相对响应因子进行定量分析.
2 结果与讨论
2.1 样品前处理方法的优化

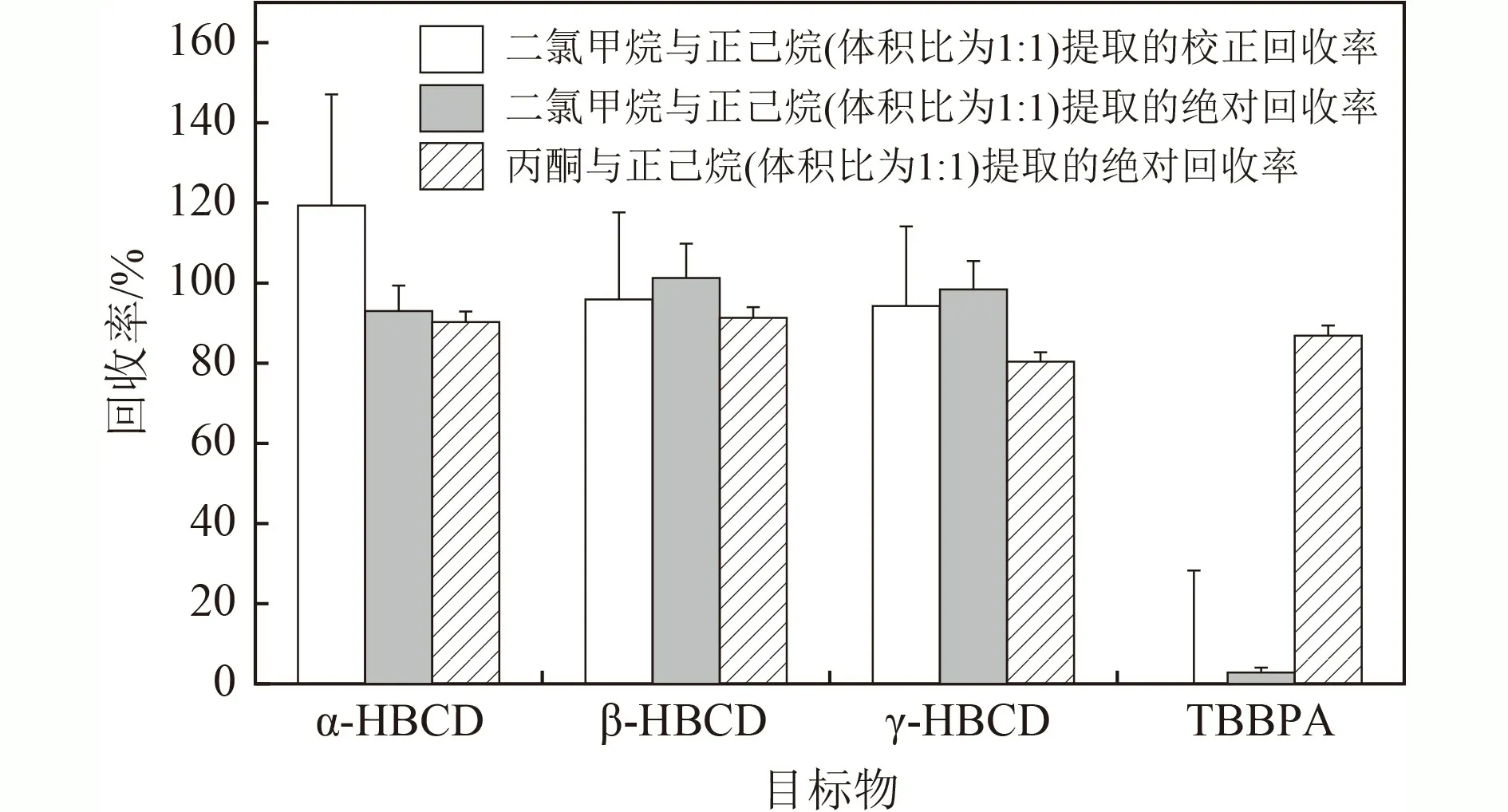
图1 不同溶剂提取目标物的回收率Fig.1 The recoveries of the analytes on different extracting solvents
有效的净化处理方式能降低基质效应,提高定性定量的准确性. 土壤和沉积物中两类目标物的净化方式主要有填充柱和固相萃取柱. 前期发现,采用单独的固相萃取小柱(C和硅胶),因柱容量和净化能力有限,净化效果不足,基质效应偏高,影响定量计算,尤其是硅酸镁小柱无法将TBBPA 洗脱下来. 为了应对复杂基质对目标物定量分析的影响,该研究采用复合硅胶柱的净化措施,首先考察了各类改性硅胶对目标物的影响. 由图2 可见,无水硫酸钠没有吸附目标物,中性硅胶和44%酸性硅胶条件下目标物的回收率为93.1%~106%,2% KOH 碱性硅胶条件下TBBPA的回收率偏低(29.4%),可能是由于碱性条件下TBBPA电离产生不可逆吸附所致. 因此,后续选择中性硅胶和酸性硅胶复合的方式对目标物进行净化处理.
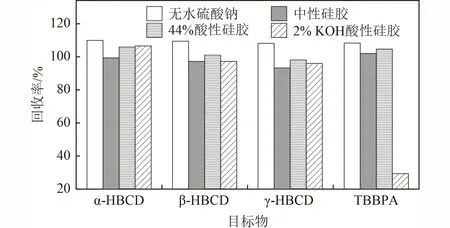
图2 目标物在4 种不同材料上的洗脱效果Fig.2 Elution results of targets on four different materials
选择复合硅胶填充柱(从下至上依次填充1 g 无水硫酸钠、1 g 中性硅胶、10 g 酸性硅胶、1 g 无水硫酸钠)进行洗脱曲线试验. 由图3 可见, 170 mL 正己烷洗脱目标物的回收率低于1%,40 mL 二氯甲烷以及80 mL 正己烷与二氯甲烷(体积比为1∶1)均能将目标物全部洗脱下来,且样品基质效应得到有效降低(低于20%). 在实际样品分析过程中,为了保证目标物的洗脱效率,需要适当增加洗脱量. 针对各类复杂基质的土壤和沉积物样品,采用中性和酸性复合硅胶填充柱作为净化手段,使用50 mL 二氯甲烷和20 mL正己烷活化,样品浓缩液上柱后,使用50 mL 正己烷淋洗净化,100 mL 正己烷与二氯甲烷(体积比为1∶1)洗脱并收集.
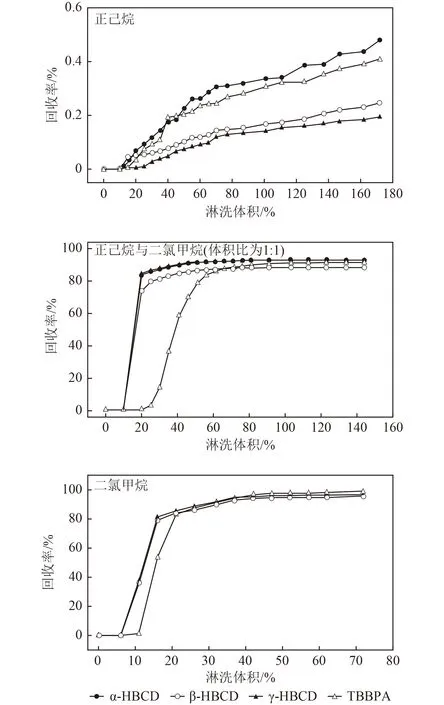
图3 目标物在3 种不同洗脱溶剂下的淋洗曲线Fig.3 The elution curves of targets in three different solvents
2.2 仪器测试条件的优化
前期试验结果显示,液相色谱系统不存在目标物的仪器背景干扰. 有关HBCDs 和TBBPA 液相色谱-串联质谱法涉及的流动相有水、甲醇、乙腈,以及三者之间不同比例的混合. 为了增加离子强度,也有在水相或有机相中添加乙酸铵,通过氨水或甲酸调节pH 的做法,但未对各条件下目标物的响应进行讨论.结合文献资料,以乙腈为有机相,考察4 种不同水相(无添加、0.05%氨水、2 mmol/L 乙酸铵和0.01%乙酸)下目标物的峰面积响应. 由图4 可见,在无添加条件下4 种目标物整体响应最高;2 mmol/L 乙酸铵条件下,HBCDs 响应与纯水的相近,但TBBPA 偏低;而0.05%氨水和0.01%乙酸条件下,4 种目标物的响应最低. 因此,选择纯水和乙腈为流动相.
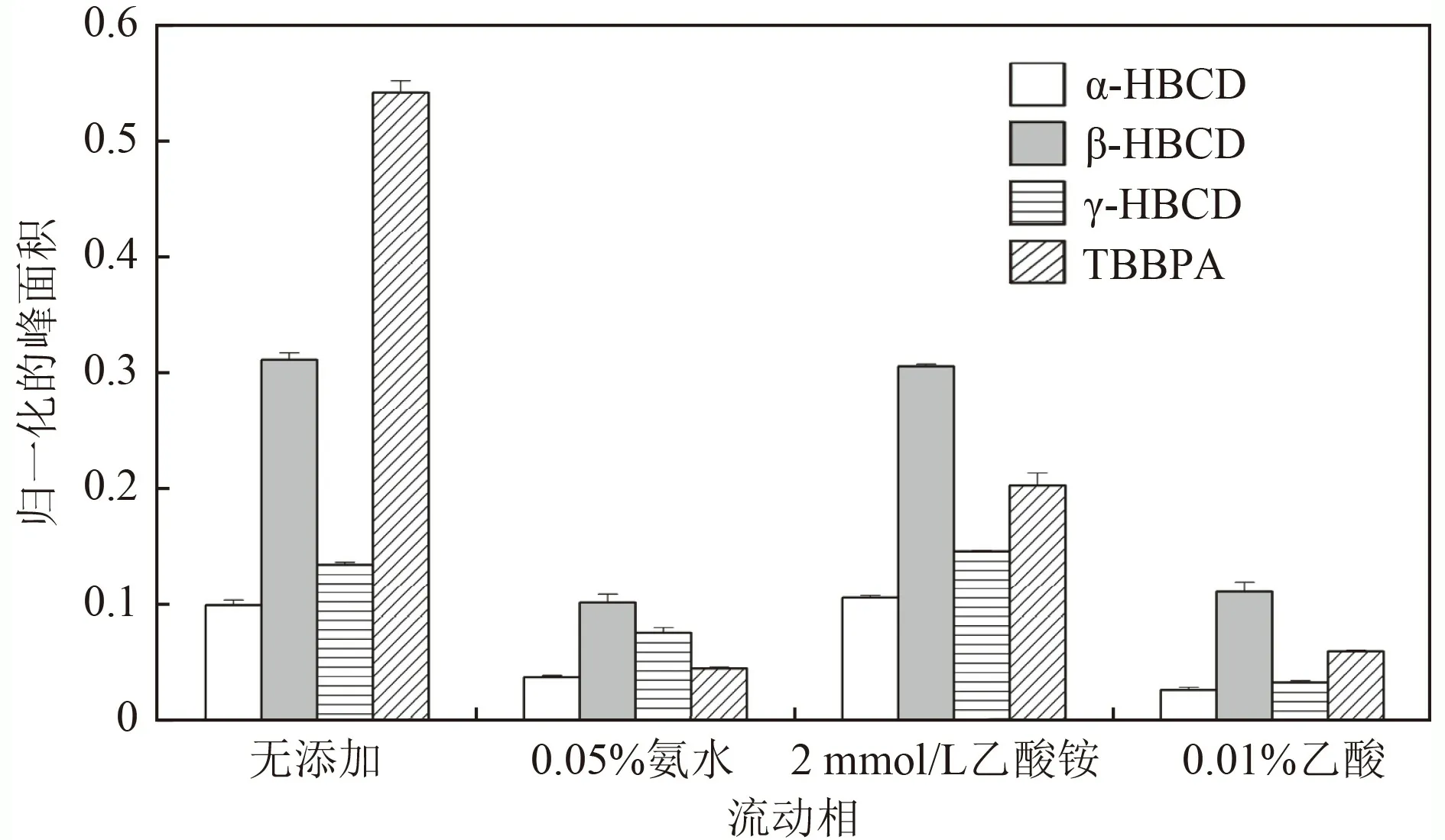
图4 不同流动相下目标物的归一化响应Fig.4 Normalized response of targets in different mobile phases
此外,有机流动相乙腈有利于β-HBCD 和γ-HBCD的分离,甲醇有利于α-HBCD 和β-HBCD 的分离. 当采用乙腈与甲醇(体积比为1∶1)的混合有机相时,3种HBCDs 异构体能够实现更大跨度的分离,该结果与已有研究结果一致. 乙腈能够实现HBCDs 异构体的基线分离,且色谱柱柱压较低,推荐采用水(流动相A)和乙腈(流动相B)作为流动相组合,参考的梯度洗脱程序如表1 所示,对应的目标物提取MRM色谱图如图5 所示.
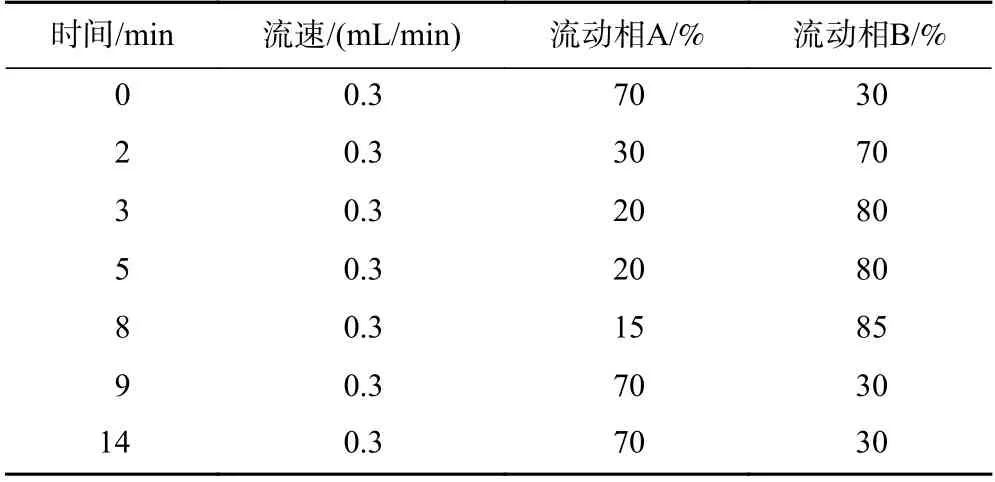
表1 目标物分析的梯度洗脱程序Table 1 Gradient elution profile for the target compounds analysis
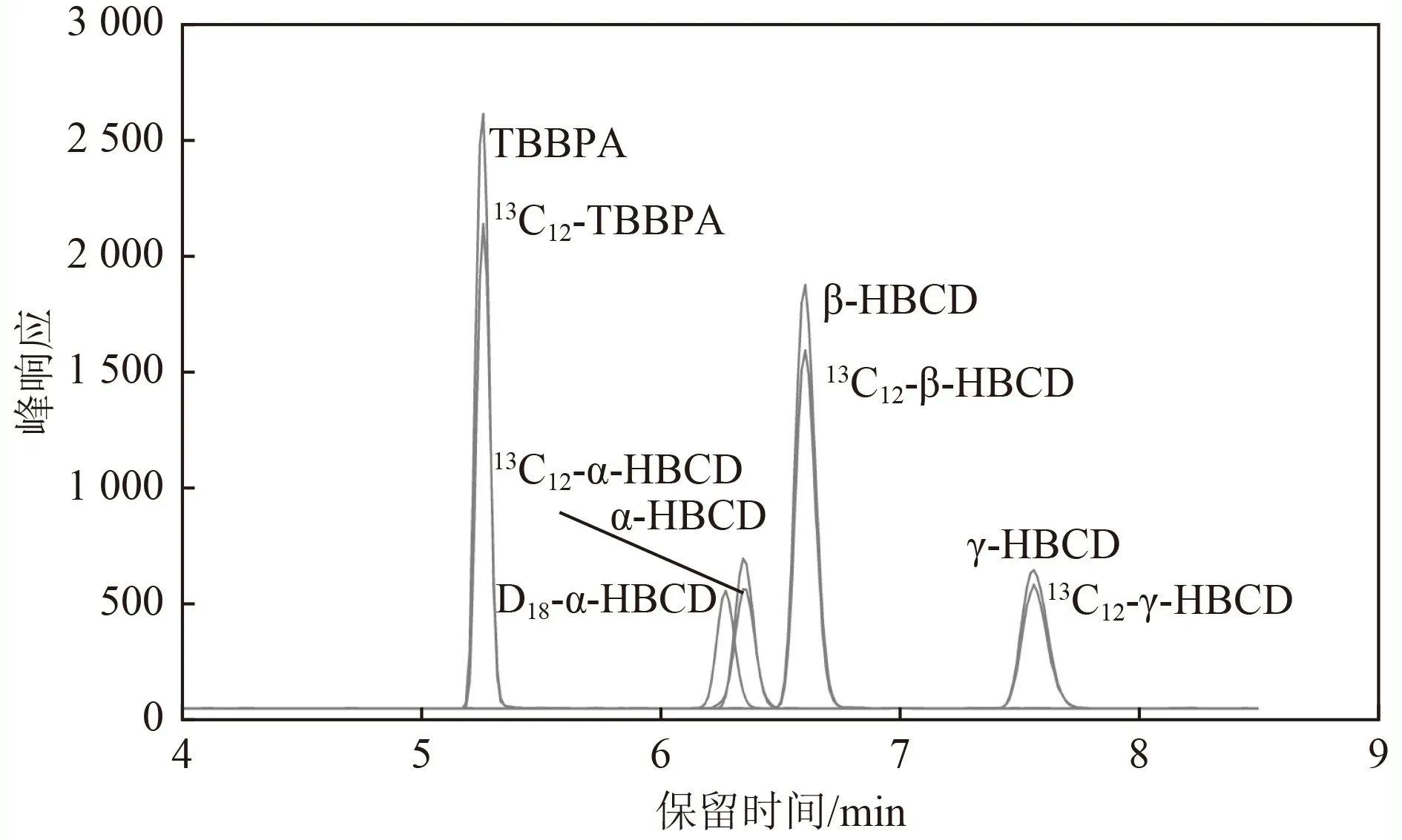
图5 HBCDs 和TBBPA 的MRM 色谱图Fig.5 MRM chromatograms of HBCDs and TBBPA
质谱参数能显著影响目标物的响应,该研究采用安捷伦液相色谱-三重四极杆质谱仪器,涉及7 个参数,且参数之间有一定的组合关系,而已有研究中少有提到对参数的全面优化. 该研究以100 ng/mL 的标准溶液为基础,在仪器参数允许范围内,设置优化范围和步长,按照顺序,逐一对7 个源参数(鞘气温度、鞘气流速、喷嘴电压、毛细管电压、雾化气、干燥气温度和干燥气流速)进行优化. 其中,鞘气温度的设定优化范围为200~380 ℃,目标物的响应随鞘气温度的升高逐渐增加,在大于340 ℃后快速降低;鞘气流速和喷嘴电压与目标物响应均呈正相关,毛细管电压在1 500 V 时,目标物响应获得最大值后迅速下降;干燥气温度的变化对TBBPA 的响应有限,干燥气流速与TBBPA 响应呈负相关,与HBCDs 呈正相关. 综合两类目标物的响应,确定的最佳离子源参数为鞘气温度340 ℃、鞘气流速11 L/min、喷嘴电压2 000 V、毛细管电压1 500 V、雾化气35 psi、干燥气温度280 ℃和干燥气流速6 L/min.
柱温影响目标物在色谱柱中的分配和分离效果,研究显示,HBCDs 和TBBPA 的柱温通常在30~40 ℃之间. 该研究从25 ℃开始对柱温进行优化,结果如图6 所示. 随着柱温的增加,目标物在色谱上的保留降低,出峰时间缩短,其中γ-HBCD 受到的影响最明显,出峰时间从柱温25 ℃时的8.5 min,缩短至42.5 ℃时的7.5 min. 出峰时间的缩短,一方面引起α-HBCD 和β-HBCD 的分离度降低,另一方面目标物在出峰时流动相中乙腈比例下降,导致目标物响应受到影响. 结果显示,35 ℃时目标物的响应值较其他柱温时偏高,且各异构体均能实现基线分离,因此选择35 ℃作为最佳柱温.

图6 不同柱温下HBCDs 和TBBPA 的MRM 色谱图Fig.6 MRM chromatograms of HBCDs and TBBPA in different column temperatures
进样量的增加能够提高方法的灵敏度,但可能影响分离和峰形. 该研究考察了进样体积1~20 μL 内目标物峰面积的变化情况. 结果显示,峰面积随进样量增加逐渐递增,但在进样量为20 μL 时偏离线性趋势,TBBPA 和α-HBCD 的变化更明显,在进样量为20 μL 时峰面积显著增加. β-HBCD 的响应与进样量之间存在二次曲线拟合(=0.999),γ-HBCD 是唯一的响应与进样量呈线性的目标物(=0.999 6). 从降低检出限角度应选择最大进样量,但进样量大于10.0 μL 后溶剂效应增强,目标物的色谱峰形出现前展驼型峰. 因此,为了保证线性范围,提高定量分析的准确性,进样量确定为5.0 μL.
采用已建立的方法,制备2.00、5.00、10.0、20.0、50.0、100 和200 ng/mL 的校准标准溶液进行分析测试,使用同位素稀释法的平均相对响应因子法进行定量计算,平均相对响应因子的相对标准偏差小于15%. 每种目标物均对应一个内标物质,采用同位素校正的方法能最大限度增加目标物定量分析的准确性. 考察连续3 d 测试样品中内标物质叠加的色谱图,评估保留时间的稳定性. 结果显示,相对于标准溶液中内标物的保留时间,样品中内标物的保留时间的相对偏差均小于2.5%. 保留时间波动最大的化合物是C-TBBPA,波动范围的差值为0.13 min. 与标准溶液中的保留时间相比,实际样品中的保留时间略微提前,可能是基质效应引起的. 从定性角度考虑,保留时间的偏差控制在±0.3 min 以内,相对偏差控制在±3.0%以内.
2.3 方法检出限和方法参数
依据《环境监测分析方法标准制订技术导则》(HJ 168-2020)附录A 有关方法检出限的计算方法,对石英砂空白基质进行7 次低浓度(0.2 μg/kg)加标试验. 测试结果求得标准偏差,基于值得到α-HBCD、β-HBCD、γ-HBCD 和TBBPA 的方法检出限分别为0.06、0.04、0.04 和0.06 μg/kg,测定下限分别为0.24、0.16、0.16 和0.24 μg/kg. 该结果满足50%的被分析物样品浓度在3~5 倍计算出的方法检出限的范围内,至少90%的被分析物样品浓度在1~10 倍计算出的方法检出限范围内的要求.
该方法检出限在0.04~0.06 μg/kg 之间,低于文献中报道的背景地区的环境浓度水平,能够满足我国履约监测的需要. 文献中的方法检出限多是基于3 倍信噪比的计算,且样品定容体积和进样量差别较大. 在统一样品分析参数的基础上,该方法的检出限与已有研究结果处于同一水平. 研究发现,沉积物中α-HBCD 检出限为0.004 μg/kg,对应的定容体积为0.2 mL,当定容体积为1 mL 时,检出限为0.02 μg/kg. 另有研究发现,沉积物中HBCDs 三个异构体的方法检出限为0.003~0.005 μg/kg,其进样体积为10 μL,定容体积为0.2 mL,换算为定容体积1 mL、进样量5 μL 时,检出限为0.03~0.05 μg/kg,与笔者研究中的方法检出限相当.
对加标浓度为0.5、2.0 和10.0 μg/kg 的石英砂样品,加标浓度均为0.5 和2.0 μg/kg 的背景土壤和海洋沉积物样品,加标浓度均为2.0 和10.0 μg/kg 的工业土壤和河流沉积物样品,各进行6 次平行测定,考察分析方法的精密度和正确度. 由表2 可见,样品加标回收的精密度为1.76%~10.6%,加标回收率(正确度)为92.9%~121%.
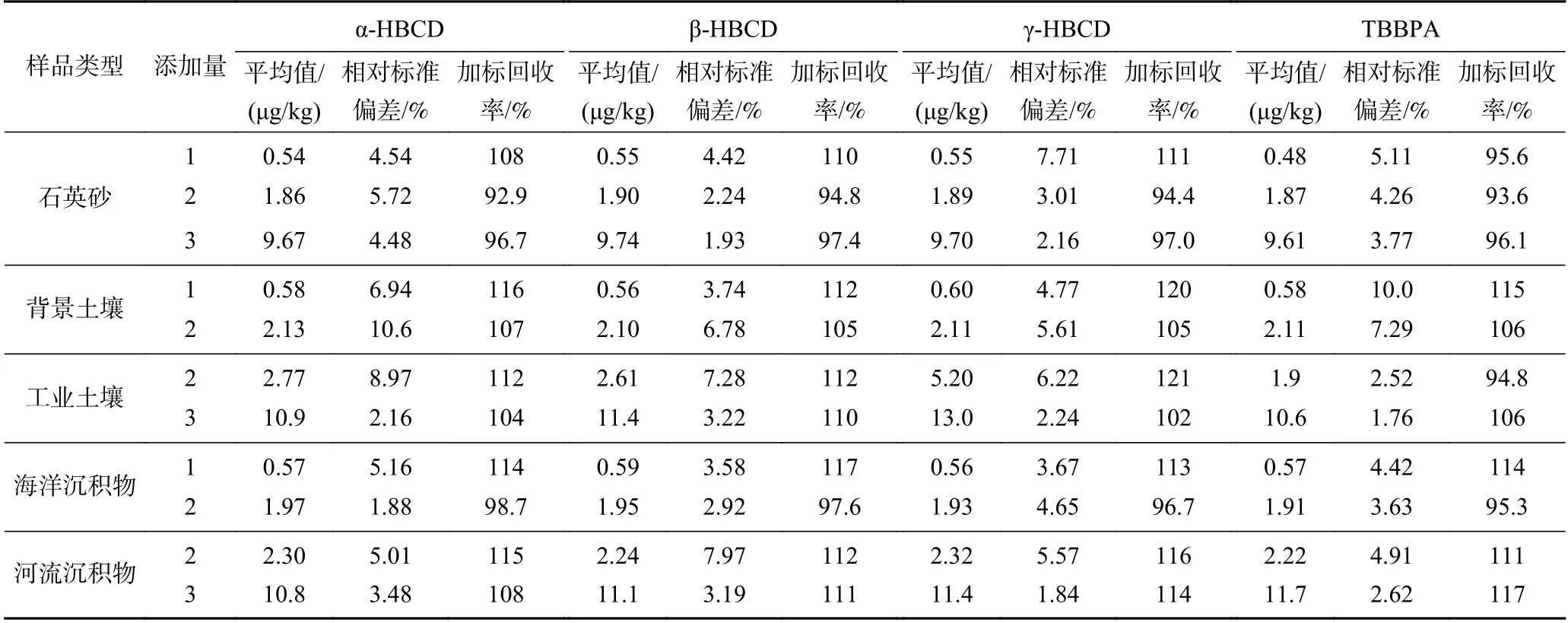
表2 方法精密度和正确度数据Table 2 The precision and accuracy of the developed method
2.4 方法适用性研究
采集8 种不同地区和类型的土壤样品,考察pH以及基质对方法适用性的影响. 土壤样品类型覆盖了我国南部、北部及中原地区,分别为湖北棕土(pH=5.54)、天津潮土(pH=8.17)、湖北棕褐土(pH=5.85)、湖北棕土(pH=5.19)、河南水稻土(pH=6.29)、云南紫色土(pH=7.51)、新疆灌淤潮土(pH=8.67)和贵州黄棕壤(pH=4.26). 对土壤样品进行3 次平行加标试验,结果显示,目标物的加标回收率范围为85.9%~104%,C-α-HBCD、C-β-HBCD、C-γ-HBCD、C-TBBPA 的回收率分别为56.5%~124%、54.0%~107%、59.1%~126%和52.2%~144%.
3 结论
a) 该研究建立了土壤和沉积物中HBCDs 和TBBPA 的高效液相色谱-三重四极杆质谱结合同位素稀释定量的分析方法.
b) 当取样量为10.00 g、定容体积为1.0 mL 时,α-HBCD、β-HBCD、γ-HBCD 和TBBPA 的方法检出限在0.04~0.06 μg/kg 之间,能够满足实际样品的分析测试要求.
c) 目标物在石英砂空白基质和实际样品的低中高浓度的加标回收率为92.9%~121%,相对标准偏差为1.76%~10.6%. 代表性土壤的加标回收率范围为85.9%~104%. 该研究建立的方法具有灵敏度高、实用性好的特点,能够适用于复杂土壤和沉积物样品中HBCDs 和TBBPA 的分析测试.
猜你喜欢
化工设计通讯(2022年10期)2022-12-31 20:42:50
波谱学杂志(2022年2期)2022-06-14 09:52:02
石油炼制与化工(2022年2期)2022-02-15 11:42:26
应用化工(2021年4期)2021-05-20 09:43:36
化工管理(2020年26期)2020-10-09 10:05:16
山东化工(2019年2期)2019-02-21 09:29:32
中成药(2017年4期)2017-05-17 06:09:46
核科学与工程(2015年3期)2015-09-26 11:58:24
现代检验医学杂志(2015年1期)2015-02-06 01:59:14
天然产物研究与开发(2014年6期)2014-04-27 14:15:54
