
Synthesis, optical properties and self-organization of blue-emitting butterfly-shaped dithienobenzosiloles
2022-09-15 03:11GaozhangGouZhaolingZhangTaoFanLeiFangMingxianLiuLiangchunLi
Chinese Chemical Letters 2022年9期
Gaozhang Gou, Zhaoling Zhang, Tao Fan, Lei Fang, Mingxian Liu, Liangchun Li
Shanghai Key Lab of Chemical Assessment and Sustainability, School of Chemical Science and Engineering, Tongji University, Shanghai 200092, China
ABSTRACT Ten novel butterfly-shaped dithienobenzosilole-based luminogens, which are peripherally installed with a variety of substituents including hydrogen, phenyl and substituted phenyl groups, have been readily prepared via an iodine-induced intramolecular electrophilic double-cyclisation reaction and subsequent deiodination or coupling reactions.The optical and electrochemical properties of these compounds were systematically investigated to clarify the relationships between their structures and properties, supported by theoretical calculations.These compounds exhibit deep-blue to sky-blue emissions and high photoluminescence quantum yields up to 0.84 in solution and solid states which are regulated by the functional blades and their steric hindrance on the α– and β–positions of thiophene rings.Their high thermal- and photo- stabilities have been revealed and mainly attributed to the dithienobenzosilole core.
Keywords:Dithienobenzosilole Luminogen Double-cyclisation Blue-emitting Photo-stability
Over the past two decades, fluorenes and polyfluorenes (PFs)derivatives have achieved great applications for the blue lightemitting diodes (OLEDs), tytically as the light-emitting active layers [1,2].However, for the future high-performanced displays, the most challenging topic remained is to realize highly stable bluecolor emission, since the typical blue-emitting fluorene derivatives have sufferred an unwanted low-energy green emission that has been attributed to the formation of carbonyl groups at the C−9 position under photo- and thermal-degradation [3].Hence,heteroatom-bridgedπ–conjugated molecules, especially siliconbridged biaryl frameworks, such as dithienosiloles and dibenzosiloles, have attracted enormous attention owing to their remarkable thermal stability and blue emissions utilised as functional organic building blocks [4–10].In particular, the dibenzosiloles have been demonstrated exhibiting high electron transport, thermal stability and excellent photoluminescence properties in virtue of the embedded metalloid Si atom and unique electronic interactions between theπ∗orbital of the conjugated modules andσ∗orbital of the exocytic silicon–carbon bonds which thereby lead to the low-lying lowest unoccupied molecular orbitals (LUMOs)[11–15].And thus the dibenzosiloles and their derivatives have been widely employed in organic optoelectronic materials, such as organic light-emitting devices (OLEDs), organic field-effect transistors (OFETs), nonlinear optic materials (NLOs), high-performance organic solar cells, fluorescence sensing and bio-imaging [16–22].Many facile and ingenious synthetic strategies to prepare dibenzosiloles have been spawned in recent years to further tailor them for novel structures and functionalities [18,23–30].
On the other hand, thiophene-incorporatedπ–extended derivatives are also particularly intriguing because of their supereminent coplanarity, charge mobility and hole-transporting properties; they are among the most successful building blocks for synthesizing highly efficient photovoltaic materials [31–35].Recently,the benzothiophene derivatives have been proved as good holetransporting materials and the integrated thiophene groups have contributed greatly to the high charge-transportation and strong luminescence [36,37].It is desirable that the integration of thiophene moieties with dibenzosiloles could generate novel materials inheriting both of their characteristics such as high thermal stability, good charge mobility and glamorous photoluminescence.Although several facile strategies, especially electrophilic iodinecyclisation and intramolecular thiolate/acetylene cyclization, were reported for the syntheses of benzothiophene and dithienofluorene derivatives [38–42], the syntheses of Si–bridged benzothiophene have been barely reported because of the very limited synthetic strategy for silole compounds and their vulnerability of acidic or basic conditions.Our group has been retaining a keen interest in the design and syntheses of dibenzosilole derivatives with intriguing structures and luminescence properties [43,44].Taking advantage of the mild iodine-induced intramolecular electrophilic cyclisation (IIIEC) reactions the difurobenzosilole derivatives could be readily synthesized in excellent yields [45].And a series of rodlike dizenzosiloles functionalized by methylthio groups have been recently reported to potentially provide superior alternatives for traditional fluorescent dyes in biological analyses and bioimaging[44,46,47].Based on these works and fascinated by the fusedthiophene and dibenzosilole compounds, we have long sought to synthesize novel dithienobenzosiloles with the concern of intrinsically molecular electronic structure, fluorescence emission and photo-stability,etc.
Herein, we report a straightforward strategy to synthesize a series of ladder-type dithienobenzosilolesπ–conjugated moduleviathe IIIEC reactions.Through the palladium-catalysed Sonogashira or Suzuki–Miyaura cross-coupling reactions versatile substituents with different bulkiness have been installed peripherally on the dithienobenzosilole core to form elegant butterflyshaped structures.Their molecular structures, intrinsic photophysical properties, thermal- and photo-stabilities as well as the molecular orbitals have been studied associated with theoretical calculations.
Ten butterfly-shaped dithienobenzosilole compounds were synthesized in three steps by the same strategy from the 2,7-dibromo-9-silafluorene (1) (Scheme 1).First of all, rigid rodlike molecules(2a–2c) containing a dibenzosilole core, ethynylene linkages and versatile aryl terminal groups were synthesizedviathe Sonogashira cross-coupling based on our previously reported method[44].With 2a–2c in the hand, the preinstalled dimethylthio groups were exploited for annulation withortho-ethynylene to construct thiophene rings onto the dibenzosilole core.It is notable that the success of the reaction depends on the electron density of the triple bonds which is affected by the peripheral substituents;this result correlates with our previous report that electrondonating groups on the ethynylene terminal are demanded for this annulation reaction [45].And then, featuring the terminal electron-donating B, C or D groups, the IIIEC reactions of 2a–2c were successfully accomplished to afford the iodine-functionalized dithienobenzosilole 3a–3c in 88.5%–90.3% yields.The fluorescence of the double-cyclisation products 3a–3c was quenched because of the introduced iodine atoms.In addition, although intermediates 3a–3c show poor solubility in common solvents, they could be treated witht-BuLi at –78 °C, followed by quenching with methanol, affording the double-deiodination compounds 4a–4c in quantitative yield.Finally, key intermediates 3a–3c as specific building blocks were found to be readily functionalizedviacoupling reactions.The substituents with different steric bulkiness were peripherally installed onto theβ–position of the thiophene ringsviathe palladium-catalysed Suzuki–Miyaura reactions to give 5a–5f in good yields.Sonogashira cross-coupling reaction of 3b smoothly proceeded to afford 6a in 71.6% yield.The efficient coupling reactions suggest that the dithienobenzosiloles 3a–3c manifest high activity for developing superior photovoltaic and photoelectric polymers as novel building blocks.All these synthesized ladder-type dithienobenzosilole compounds were fully characterised by multinuclear (1H,13C and29Si) NMR, HRMS and IR spectroscopies.
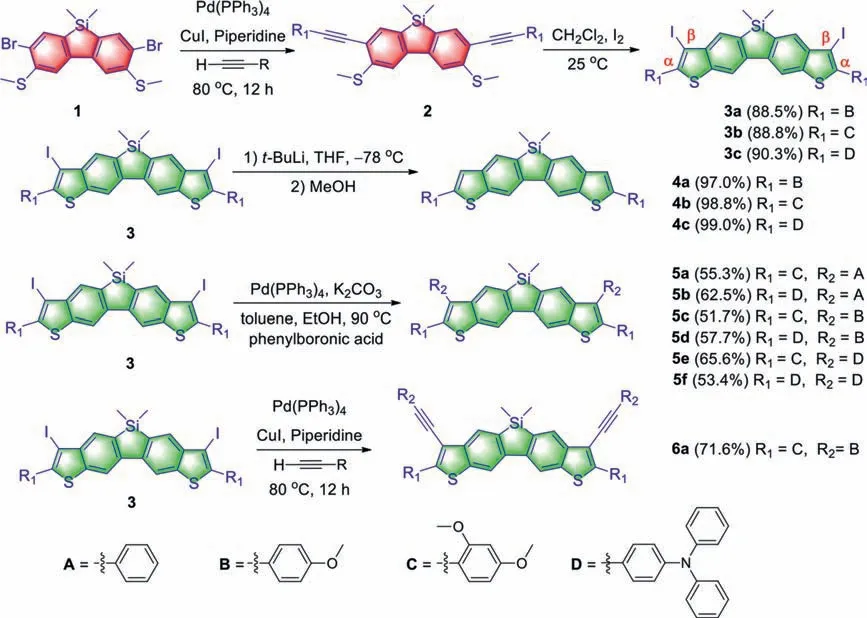
Scheme 1.Synthetic routes and chemical structures of the dithienobenzosilole derivatives.
A plausible mechanism of this IIIEC reaction is shown in Fig.S1 (Supporting information).First, molecular iodine reacts with electron-rich alkynes and forms an iodine-ethynylene complex.Thereafter, the intermediate I, I′or I′′is generated by the iodinecyclisation and the intramolecular isomerisation [48,49].The terminal electron-donating groups such as methoxy and substituted amino groups play an indispensable role in the formation of intermediate I′, which facilitated by the intramolecular attack of ortho methylthio groups could form the five-membered ring intermediate I′′.Finally, the methyl groups will be removed by I−in the systemviathe SN2reaction to generate the target double-cyclisation product.
To elucidate the molecular structures, suitable crystals of 3a,5a and 5d were grown from a mixed solution of ethanol and dichloromethane at room temperature for the X–ray diffraction analysis.The crystal structures of 3a, 5a and 5d are depicted in Fig.1 and Fig.S2 (Supporting information), and the detailed crystallographic data are summarised in Table S1 (Supporting information).The crystal packing diagrams of 3a, 5a and 5d are shown in Fig.S3 (Supporting information).Compounds 3a, 5a and 5d are crystallized in a triclinic lattice withP1space group.All the structures intuitively feature the dithienobenzosilole cores and reveal distorted tetrahedron coordination geometry around the Si atoms.The distances of the Si–C(Ar) bonds of 3a, 5a and 5d were comparable, that are, (1.874, 1.868), (1.869, 1.865) and (1.875, 1.876) ˚A,respectively.The C(Ar)–Si–C(Ar) bond angles around the respective Si atom (91.83°, 91.10° and 91.53°) correlate well with the reported dimethylthio-modified rigid rod-like dibenzosiloles, suggesting that the post-annulation reactions have little effect on the parent dibenzosilole cores [44].
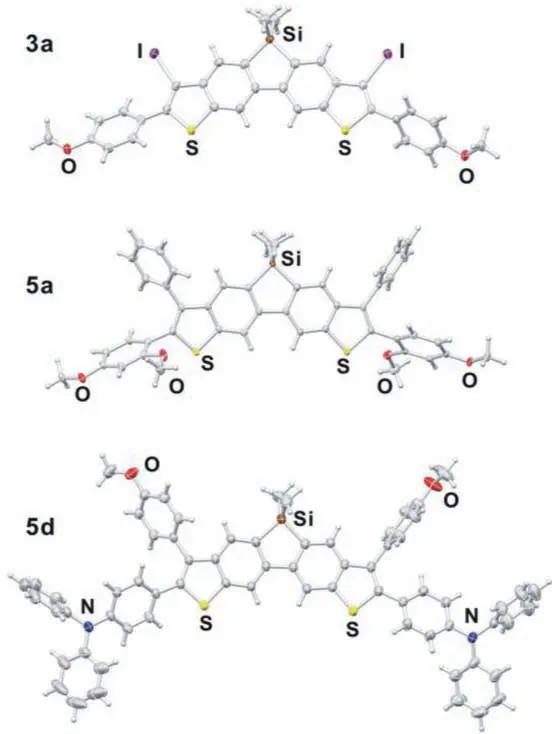
Fig.1.Crystal ORTEP diagrams of 3a, 5a and 5d
Fig.S2 shows the torsion angles between the dithienobenzosilole plane and phenyl rings on theα– orβ–positions of thiophene units which are greatly induced by the steric hindrance between the two neighbouring rings.The torsion angles of theα–positions are asymmetrically (57.36°, 39.77°), (60.58°, 74.91°)and (35.29°, 26.39°) for 3a, 5a and 5d, respectively; those of theβ–positions are (65.63°, 55.05°) and (54.03°, 80.65°) for 5a and 5d, respectively.These torsions largely influence the extent ofπ–conjugation, and thus exert great impact on the distribution ofπelectrons, photophysical properties.In addition, the steric substituents on thiophene rings and sp3–hybridised Si atom shall cause inefficient packing in the solid state [7].The crystal packing diagrams reveal that these compounds indeed exhibit ineffectiveπ–πstacking interactions.The distances between the layers are 4.12 and 6.55 ˚A for 3a and 5a respectively, while compound 5d exhibits the largest packing distance accompanied by slight bending of the dithienobenzosilole core (Fig.S3).
Figs.S4–S7 (Supporting information) show the absorption and fluorescence spectra of 4a–6a, and the data are collected in Table S2.The UV–vis absorption spectra inn-hexane and THF are shown in Figs.2a and c respectively, and the corresponding photophysical data are listed in Table 1.All the compounds exhibit absorption maxima at 331–406 nm with very large extinction coefficients (2.66–9.66 × 104L mol−1cm−1) and intense absorptions in the range of 296–402 nm (2.26–9.94 × 104L mol−1cm−1).The low-energy absorption bands, which are significantly split into two peaks, could be assinable to the 0→0 vibrational bands of the strong S0→S1transition due to the newly formed thiophene rings.Moreover, no discernible red/blue shift accompanied by distinct solvent polarity effect is observed, indicating that the absence of intramolecular charge transition (ICT) with proper electron-withdrawing characteristic of the dithienobenzosilole core(Figs.S4–S7).
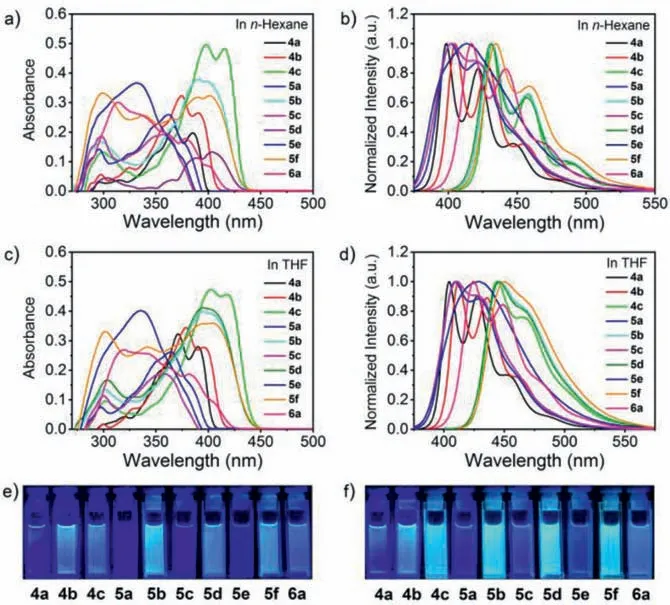
Fig.2.UV–vis absorption and emission spectra of 4a–6a in n-hexane (a, b) and tetrahydrofuran (THF) (c, d) measured at a concentration of 5 × 10−6 mol/L.Photographs of 4a–6a captured in n-hexane (e) and THF (f) under UV light (365 nm).
Among these compounds, 4b, 5a, 5c and 5e with the peripheral substituents on theβ–positions of the thiophene rings from H,phenyl andp-methoxyphenyl to diphenylaminophenyl (DPAP) exhibit significant blue shifts from 390 nm to 331 nm inn-hexane solution.Similiarly, the same phenomenon was observed for the analogous compounds, 4c, 5b, 5d and 5f in which the same DPAP group is installed on theα–positions.With the same substituent on theβ–positions, varying the substituents on theα–positions from dimethoxyphenyl to DPAP results in a significant red-shift in the absorption maximum and broader absorption bands such as 5e versus 5f.Compared with analogue 4b, compound 6a shows a broad band and hypsochromic shift in the absorption.It was found that electron-donating orπ–conjugated substituents on theα–position of the thiophene rings could cause the red-shift in the absorption, while the introduction of the steric bulky substituents on theβ–positions resulted in a blue shift.Thus, the absorption spectra of this system can be jointly tailored by the substituents on theα– andβ–positions of the thiophene rings.
The emission spectra inn-hexane and THF are shown in Figs.2b and d.Upon excitation at the absorption maxima, all the dithienobenzosilole compounds (4a–6a) exhibit intense emissions withλmaxof 398–458 nm corresponding to deep to sky blue emissions in solution, as well as 11%–84% quantum yields (QYs) in the air at an ambient temperature (Figs.2e, f, Figs.S8–S12 in Supporting information and Table 1).The emission spectra of 4a and 6a inn-hexane feature broad bands withλmax(lifetime, QY) of 421 nm (τ= 0.44 ns, 69%) and 441 nm (τ= 0.44 ns, 31%), respectively.Comparatively, in THF solution, a little bathochromic shift,improved lifetime and QY are obtained,i.e., 427 nm (τ= 0.52 ns,77%) for 4a and 448 nm (τ= 0.51 ns, 43%) for 6a.Moreover,the emission spectra of 4b, 5a, 5c and 5e inn-hexane manifest broad bands withλmax(lifetime, QY) of 402–428 nm (τ= 0.36–1.17 ns, 11%–79%) and 409–435 nm (τ= 0.48–1.00 ns, 9%–82%)in THF.In particular, in the same solvent (e.g., n-hexane), the QYs of 4b, 5a, 5c and 5e with the increase of steric hindrance on theβ–position of the thiophene rings significantly decrease from 0.75 for 4b to 0.11 for 5e.The emission spectra of 4c, 5b, 5d and 5f inn-hexane feature broad bands withλmax(lifetime, QY) of 434–458 nm (τ= 0.45–0.52 ns, 49%–78%) and 444–449 nm (τ= 0.52–0.62 ns, 58%–79%) in THF.For the compound series of 4c, 5b, 5d and 5f, the variation tendency of QY andλmaxis similar, that is, a certain decrease with some blue shift.
To gain a deeper insight into the correlation between the photophysical property and molecular structure, the whole moleculardistortions are attempted to be evaluated by the sum of the torsion angles (Sϕ) of the substituents on theα– andβ–positions of the thiophene rings.By analysis of the crystal structures, theSϕof 5a is much larger than those of 3a and 5d.The other compounds without crystal structures are verified from the optimised structures by density-functional theory (DFT) calculations at the B3LYP/6–31+G(d, p) level (Fig.S13 in Supporting information).It was found that the QY (ΦF) in solution is particularly affected by intramolecular rotation especially from the steric hindrances on theα– andβ–positions.The extents of the intramolecular rotation interfered by the steric hindrance could result in sharp distinction between the radiation (kr) and non-radiation (knr) rate constants,thus greatly affecting the QY (Table 1).The largeSϕvalues, which could estimate the degree of intramolecular rotation, will cause radiation-less relaxation and further affect QYs and spectra.In general, the installed electron-donating orπ–conjugated substituents on theα–positions of the thiophene rings lead to red-shift in spectra, whereas introduction of the substituents with steric bulkiness on theβ–positions result in blue-shift in spectra and decreased QY.This rule revealed here is of great significance in applying molecular engineering for the design and syntheses of dithienobenzosilole derivatives with adjustable absorption and emission, as well as improved QYs.
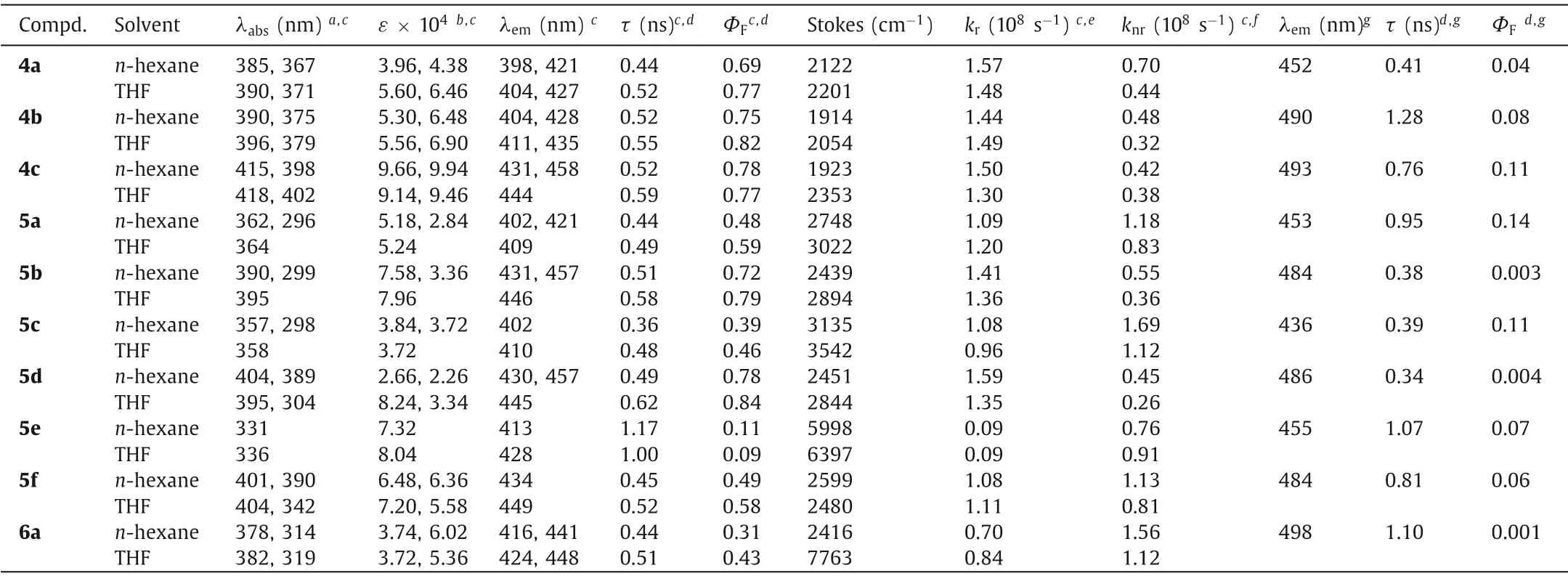
Table 1 Selected photophysical data of 4a–6a in different solvents at 298 K (c = 5 × 10−6 mol/L).
For this kind ofπ–conjugated molecules installed with aryl blades, their photophysical variations in various states are particularly interesting, since a phenomenon antithetic to the general aggregation-induced quenching (ACQ) has been reported and termed aggregation-induced emission (AIE) [50–53].It has been revealed that the main contributions to the AIE effect are the restricted intramolecular rotation and suppressedπ–πstacking by the twisted molecular conformation of AIEgens in the aggregation state.And thus the energy of the excited molecules will be overwhelmingly exhausted by the radiative channel leading to enhanced emission [54,55].The photophysical properties of 5a–6a were investigated by their fluorescence spectra in THF/H2O mixtures with different water fractions (fw) in a concentration of 10 μmol/L (Figs.S14–S17 in Supporting information).Compounds 5a–6a exhibited strong fluorescence in the THF solution but the emission intensity gradually decreased and the emission maxima are red-shifted withfwincreasing, which could be classified as a typical ACQ effect.This is because 5a–6a as a novel molecular system featuring largeπ–conjugated dithienobenzosilole core and plenty of peripheral blade groups are likely to have impededπ–πstacking but the intramolecular dynamics cannot be completely restricted in the aggregation state [56].
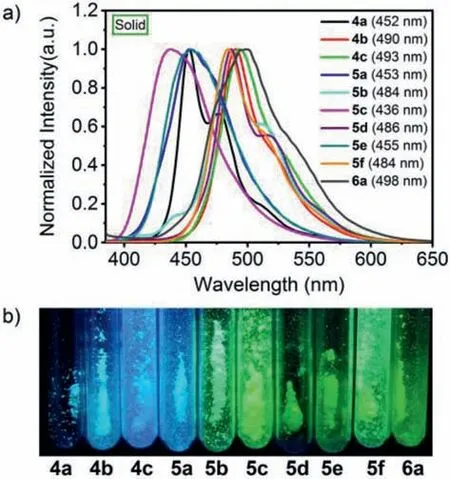
Fig.3.(a) Normalized solid-state emission spectra of 4a–6a measured at room temperature.Inset: emission maximum.(b) Photographs of compounds 4a–6a powders under 365 nm irradiation.
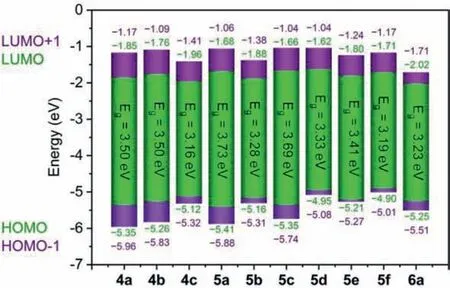
Fig.4.Energy level diagrams of the frontier orbitals of 4a–6a by DFT calculations.
In view of that various applications of the organic emitting materials have been developed in their solid state, the solid-state photophysical properties of 4a–6a were studied systematically (Fig.3 and Table 1).Owing to the dithienobenzosilole core, the normalised solid-state emission maxima of 4a–6a cover a wide range from 436 nm to 498 nm.The emission maxima of all the compounds are red-shifted with respect to the corresponding emission maxima in solution, whereas the fluorescence QYs considerably decrease to 0.1%–11% due to the ACQ effect.Notably, the CIE color coordinates of these dithienobenzosiloles in solution are located at the blue (0.15, 0.06) or deep-blue (0.15, 0.10) light emission region, while the powders’CIE color coordinates fall in the blue (0.15,0.06) or green (0.3, 0.6) light emission region with good and stable color purity (Figs.S18–S20 in Supporting information).Therefore,abundant alternatives to the traditional blue-emitting materials are supplied for developing this kind of key materials with high stability and color purity.
The electrochemical measurements of the dithienobenzosilolebased luminogens were conductedviacyclic voltammetry (CV)in dichloromethane (DCM) solutions utilizingn-Bu4NPF6as the supporting electrolyte to evaluate the energy levels [57].The CV curves are shown in Fig.S21 (Supporting information) and the electrochemical data, calculated highest occupied molecular orbit (HOMO) and lowest unoccupied MO (LUMO) energy levels are summarised in Table S3 (Supporting information).The oxidation peaks of the dithienobenzosilole luminogens are calculated by the onset values to be 1.17–1.94 V.The HOMO energy levels are estimated from the oxidation potential according to equationEHOMO= −(Eonset+ 4.74) eV, and then the LUMO energy levels(ELUMO=EHOMO+Eg) are determined by the optical bandgaps.Compounds 4a–6a exhibit the first irreversible oxidation onset peaks from 0.65 to 0.93 V, however the reduction waves are structureless and explicit, which could be attributed to the irreversible single-electron oxidation and reduction from theσ∗(Si)–π∗(C) interaction in the Si–bridged species [58].The corresponding electrochemical gaps fall in the range of 2.76–3.12 eV.The electrondonating ability andπ–conjugation degree of the substituents on theα–positions, as well as the steric hindrance of the substituents on theβ–positions significantly affect the bandgaps, as observed from the optical and electrochemical measurements.
To gain insight into the geometric, frontier orbital (FO), electronic and spectroscopic properties of the dithienobenzosiloles,DFT and time-dependent DFT (TD–DFT) calculations were conducted with the Gaussian 09 package [59].The geometric structures were optimised, employing the B3LYP/6–31+G(d, p) basis set with single-point energy.The UV–vis spectra and MOs were calculated with the TD–DFT (SCRF(PCM/Bader)–B3LYP/6–31+G(d, p)) basis set.The results of electronic absorption spectroscopy are shown in Figs.S22 and S23 (Supporting information) and the data are summarized in Tables S4 and S5 (Supporting information), which are well consistent with the experimental data.The minimum deviation between the calculated and experimental values of the absorption maxima is only 0.54%, namely the vertical excitation to the first excited state primarily involving the HOMO to LUMO(π→π∗) transition (>94%) with very high oscillator strength.The FO energy level diagrams of 4a–6a are shown in Fig.4.The contours of MOs (HOMO−1, HOMO, LUMO and LUMO+1), along with the electrostatic potential maps, are shown in Figs.S24 and S25(Supporting information).The calculated HOMO and LUMO levels are in the range from –5.41/–4.90 to –2.02/–1.62 eV.Therefore,the theoretical energy gaps (Eg) of 4a–6a are 3.50, 3.50, 3.16, 3.73,3.28, 3.69, 3.33, 3.41, 3.19 and 3.23 eV.According to Fig.S24 and S25, HOMO and LUMO of 4a, 4b, 4c, 5a, 5c and 6a are distributed in the dithienobenzosilole cores, indicating that the S0→S1transitions of these compounds exhibit a local excitation characteristic.In addition, HOMOs of 5b, 5d, 5e and 5f are dominated on the dithienobenzosilole core and the substituent on theα–positions of the thiophene rings, while the LUMO densities are mainly concentrated on the dithienobenzosilole moieties, indicating the latent occurrence of the intramolecular charge transfer process.The calculatedEgvalues are larger than the experimental values from CV and the UV–vis absorption spectra but the trends exhibit a good coincidence.
The thermal properties of the dithienobenzosilole luminogens were verified by the fluorescence spectra of the 4a–6a powder samples annealed in the air for 15 h at 200 °C in comparison with their spectra of pristine samples.Fig.5 shows the normalised fluorescence spectra of the pristine and annealed 4a–6a.No new emission bands, in particular the unwanted low-energy green emissions as that of the fluorenes, were detected for the annealed samples; each annealed sample exhibited identical spectrum to that of the corresponding pristine material, suggesting that the butterflyshaped dithienobenzosiloles possess good thermal stability [60].In addition, thermogravimetric analysis (TGA) of the representative compounds 4c, 5b and 5f were conducted under nitrogen atmosphere.The decomposition temperatures (Td) estimated by 5%weight loss were 345, 470 and 498 °C respectively, also suggesting the high thermal stability of these butterfly-shaped dithienobenzosiloles (Fig.S26 in Supporting information).
The structures of these butterfly-shaped dithienobenzosiloles are similar to those of diphenylindene (DPI) derivatives [61],which exhibits extremely poor photo-stability on account of the reversible photo-cyclisation (PC).Other derivatives including spirofunctionalized diphenylethenes [62], tetraphenylethene (TPE)derivatives [63], diarylethenes (DAEs) [64],etc.also display unsatisfactory photo-stability ascribed to a similar photochromism reactivity.The photostability of luminogens is of vital importance in consideration of various applications involving light radiation.To our delight, except for the outstanding thermal stability, the dibenzosilole derivatives also manifest compelling photo-stability.
The photo-stability of 5a–5f were tested in an argon-saturated DCM solution (5.0 × 10−6mol/L) by tracking the variations of UV–vis and fluorescence spectra after UV exposure from a lamp at 365 nm (power density = 86.5 mW/cm2) under ambient temperature.The absorption and fluorescence spectra of 5a–5f after UV exposure and the plots of the relative emission intensity (F/F0) versus the relative absorbance intensity (Abs/Abs0) are shown in Fig.S27 and S28 (Supporting information).Compounds 5a–5f show good to excellent photo-stability after UV light irradiation for 60 or 90 min.About 80% and 60% of the fluorescence intensity is retained for 5a and 5c after the exposures for 60 and 90 min respectively, while negligible changes in the absorption and emission intensity are observed for the other compounds.These results indicate that introduction of the amino group DPAP could considerably improve the photo-stability of dithienobenzosilole-based luminogens.Additionally, the photo-stability of 5a–5f has been monitored by taking the1H NMR spectra (1.0 mg in 0.6 mL of C6D6or CDCl3) under 365 nm irradiation in argon atmosphere at ambient temperature.As shown by the time-dependent1H NMR spectra (Figs.S29–S34 in Supporting information), no discernible variation appears for all these compounds after UV light irradiation for 8 h.Representative compounds 4c, 5b, 5d and 5f even show persistent photo-stability under the UV irradiation in the air-saturated solution (Figs.S35 and S36 in Supporting information).Compared with our previously reported rigid rod-like dimethylthio-modified dibenzosiloles[44], the photo-stability, photo-oxidation stability and QYs of these butterfly-shaped dithienobenzosilole-based luminogens have been greatly improved, suggesting that annulationviathis IIIEC reaction is a straightforward strategy to not only modulate the photophysical properties but also enhance the photo-stability and photooxidation stability [65,66].
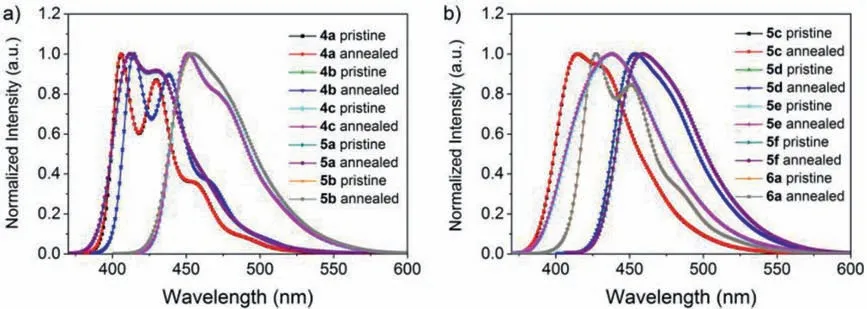
Fig.5.Fluorescence spectra of pristine and annealed 4a–6a in DCM.

Fig.6.SEM images of self-assembled 4a in toluene (a, b) and 5f in n-hexane (c–e).
Compounds 4a and 5f were selected as representatives for selforganization in verious solvents, such asn-hexane, toluene and DCM/n-hexane (1:1),viaslow evaporation method (Fig.6 and Figs.S37–S40 in Supporting information).In particular, both of them exhibit a strong tendency to self-organize into different morphologies from different solvents and were characterised by scanning electron microscopy (SEM).As shown in Fig.6, 4a and 5f are readily to self-organize into one-dimensional micro-sheets and sticks which possess great potential in the applications of electronic devices such as photonic crystal and OFETs.
In summary, ten novel butterfly-shaped dithienobenzosilolebased luminogens were synthesizedviathe iodine-induced intramolecular electrophilic double-cyclisation reaction.The readily produced diiodide dithienobenzosilole can serve as a useful building block for constructing versatile optoelectronic materials through the practicable cross-coupling reactions.Their photophysical and electrochemical properties, as well as thermal stability and photo-stability, were particularly evaluated and further elucidated by DFT studies.These dithienobenzosilole-based luminogens exhibit deep-blue to sky-blue emissions with very high quantum yields up to 0.84 in solution.The quantum yields of this molecular system are regulated by the substituents on theα– andβ–positions of the thiophene rings displaying different extent of intramolecular rotation based on the steric hindrances.Bulky substituents on theβ–positions result in twisted structure and distinct blue-shift in the adsorption.More rotatable and dynamic groups installed on the periphery will exhaust the excited energy in non-radiative way, leading to decreased quantum yields.These dithienobenzosiloles exhibit high thermal stability up to 200 °C and outstanding photo-stability as well as photo-oxidation stability attributed to the dithienobenzosilole core.The introduction of diphenylaminophenyl groups could suppress photo-bleaching to improve the photo-stability.The facile strategy to synthesize dithienobenzosiloles and principles for molecular engineering unveiled in this work will provide valuable guidance for molecular design and synthesis of novel organic optoelectronic materials with prominent properties such as luminous efficacy and stability in practical applications.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was financially supported by the National Natural Science Foundation of China (No.21501135), the Fundamental Research Funds for the Central Universities, and the Recruitment Program of Global Experts of China.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2021.12.052.
 Chinese Chemical Letters2022年9期
Chinese Chemical Letters2022年9期
- Chinese Chemical Letters的其它文章
- A review on recent advances in hydrogen peroxide electrochemical sensors for applications in cell detection
- Rational design of nanocarriers for mitochondria-targeted drug delivery
- Emerging landscapes of nanosystems based on pre-metastatic microenvironment for cancer theranostics
- Radiotherapy assisted with biomaterials to trigger antitumor immunity
- Development of environment-insensitive and highly emissive BODIPYs via installation of N,N’-dialkylsubstituted amide at meso position
- Programmed polymersomes with spatio-temporal delivery of antigen and dual-adjuvants for efficient dendritic cells-based cancer immunotherapy