
Programmed polymersomes with spatio-temporal delivery of antigen and dual-adjuvants for efficient dendritic cells-based cancer immunotherapy
2022-09-15 03:10HanyongWangXinyuYangChunyanHuChenluHuangHaiWangDunwanZhuLinhuaZhang
Chinese Chemical Letters 2022年9期
Hanyong Wang, Xinyu Yang, Chunyan Hu, Chenlu Huang, Hai Wang, Dunwan Zhu,Linhua Zhang
Tianjin Key Laboratory of Biomedical Materials, Key Laboratory of Biomaterials and Nanotechnology for Cancer Immunotherapy, Institute of Biomedical Engineering, Chinese Academy of Medical Sciences and Peking Union Medical College, Tianjin 300192, China
ABSTRACT Since antigen and adjuvant are rapid clearance in vivo, insufficient delivery to induce dendritic cells (DCs)maturation and cross-presentation, as well as limited migration efficiency of DCs to secondary lymph organs, greatly hinders the development of DCs-based immunotherapy.Herein, PCL-PEG-PCL polymersomes(PCEP-PS) as antigen and adjuvants delivery nanoplatforms (IMO-PS) were well-designed, which can electrostatically adsorb OVA antigen on the surface via DOTAP lipid and effectively encapsulate OVA antigen into the inner hydrophilic cavity to achieve both initial antigen exposure as well as slow and sustained antigen release, incorporate MPLA within the lipid layer to ligate with extracellular TLR4 of DCs as well as encapsulate IMQ in the hydrophobic membrane to ligate with intracellular TLR7/8 of DCs for activating synergistic immune responses via different signaling pathways.The IMO-PS significantly improved antigen uptake, promoted DCs maturation and cytokines production.DCs treated with IMO-PS could enhance migration into draining lymphoid nodes, and eventually induced antigen-specific CD8+ and CD4+T cell responses and OVA-specific cytotoxic T lymphocyte (CTL) responses.Prophylactic vaccination of EG7-OVA tumor-bearing mice by IMO-PS+DCs significantly extended tumor-free time, effectively suppressed tumor growth, and greatly extended median survival time.The strategy may provide an effective nanoplatform for co-delivery antigen and dual-adjuvants in a spatio-temporally programmed manner for DC-based cancer immunotherapy.
Keywords:DCs vaccine Immunotherapy Imiquimod MPLA DOTAP OVA
As professional antigen-presenting cells, dendritic cells (DCs)play pivotal roles in both innate and adaptive immune responses[1].By processing captured antigen into peptides, DCs present them to lymphoid organs to initiate T cell or B cell immunity [2–4].Recently, DCs have been extensively used in developing DCsbased immunotherapy to treat cancer [5].DCs-based therapy includesin vitroloading of DCs with tumor or pathogen-associated antigens, activating and maturing the cells, and then reinjecting the DCs back to activate T cellsin vivo[6–8].Although DCs-based immunotherapy exhibits safety and immunogenicity, clinical responses have been largely unsatisfactory [9].There are several pitfalls restricting the effects of DCs vaccines.Among them, the main pitfall is few (∼0.5%–2%) injected DCs can migrate to the lymph nodes (LNs), which hinders effective induction of T cell responses.Another drawback is inefficient stimulation of DCs to induce their maturation for the subsequent antigen-presentation to T cells, which is essential for effective DCs-based immunotherapy[1].
Nanotechnology offers unique opportunities to tackle the clinical challenges faced by current DCs-based immunotherapy.Nanoparticles can be designed to act on DCs and regulate their functions in several distinct ways: by delivering or acting as adjuvants to optimize DCs activation, enhancing antigen internalization by targeting to DCs, facilitating exogenous antigen crosspresentationvialysosome escape, incorporating different vaccine components to help DCs to govern the activation of CD4+and CD8+T cells, or exerting intrinsic immunomodulation effect on DCs [8,10–12].Rincon-Restrepoet al.demonstrated that antigen encapsulated within the nanoparticles preferred CD4+T cell activation while antigen decorated onto the surface of NPs primarily led to CD8+T cell responses [13].Hence, nanoparticles with both surface-attached and core-encapsulated antigen might result in both CD4+and CD8+immune responses.Among various nanoparticles, polymersomes have a unique structure in which an aqueous core is enveloped by a bilayer membrane formulated by amphiphilic copolymers, entitling the vesicles with the capability of encapsulating both hydrophobic and hydrophilic molecules[14].The potential to encapsulate bioactive molecules with various physicochemical properties makes polymersomes a flexible vehicle to incorporate both antigen and adjuvants.In addition,the surface of the polymersomes can be easily further decorated with cationic lipid, which can complex with anionic peptides or proteins to generate immune responsesin vivo.1,2-dioleoyl-3-trimethylammonium-propane (DOTAP) has been widely investigated in forming cationic liposome.DOTAP not only can adsorb anionic antigen to form vaccine carrier but also can facilitate antigen escape from the lysosome into the cytosolviaits ‘proton sponge’effect in acidic condition [15,16].Only after lysosome escape can the antigen be presentedviaMHC Ⅰpathway to activate CD8+T cells and elicit cytotoxic T-lymphocytes (CTL) response.
Many studies demonstrated that immature DCs can be effectively activated by toll like receptor (TLR) agonists, leading to their maturation and migration to the draining LNs, where they can stimulate effector T cells [17,18].In addition, activation of TLRs on immune cells can promote the secretion of pro-inflammatory cytokines such as IL-6, IL-12 and TNF-α, leading to enhanced innate and adaptive responses [19,20].TLR family members can be divided into endosome-associated and cell-membrane-associated receptors [21].R837 (imiquimod, IMQ) is an endosome-associated agonist for TLR7 and TLR8, which can activate DCs and induce the secretion of various pro-inflammatory factors [22].Monophosphoryl lipid A (MPLA), a detoxified form of lipopolysaccharide (LPS),can interact with membrane-associated TLR4 on DCs and activate DCs.MPLA has been repeatedly shown to enhance the release of pro-inflammatory cytokines and induce stronger T helper 1 (Th1)-biased immune responses [23,24].TLR7/8 signaling depends on myeloid differentiation primary-response gene 88 (MyD88) pathways, while TLR4 signaling is dependent on both of the My88 and TIR-domain-containing adaptor protein inducing IFN-β(TRIF) pathways [25,26].Activation of multiple signaling pathways from both the cell surface and endosome could spatially and temporally induce immune responses that are synergistic [27].TLR7/8 and TLR4 co-stimulation has been reported to significantly increase IL-12 secretion and skew DCs to improve Th1-biased responses [28,29].
Based on the hypothesis that the immunological efficacy of lipid-polymeric hybrid NPs can be enhancedviaincorporating appropriate molecules, this study aims to compare whether the combination of both endosome-associated and cell-membraneassociated TLR agonist can achieve the highest efficacy in DCsbased immunotherapy.OVA-loaded polymersomes containing both TLR agonist MPLA and IMQ (IMO-PS), single agonist (MO-PS and IO-PS), or without agonist (O-PS) were fabricated and characterized in terms of size and size distribution, zeta potential, and OVA loading amounts.Then, immature DCs were treated with these OVAloaded polymersomes to obtain DCs-based vaccine.The cell viability, cellular uptake and effect of various OVA-loaded polymersomes on DCs maturation, cytokine secretion, and antigen crosspresentation were evaluated.In addition,in vivotracking study was carried out to evaluate the homing ability of DCs-based vaccine.Furthermore,in vivointracellular IFN-γproduction, OVA-specific CTL responses, and prophylactic vaccination effect of various DCsbased vaccines against EG7-OVA tumor model were assessed.Our study revealed that IMO-PS stimulated DCs were more efficient in cancer immunotherapy than MO-PS or IO-PS stimulated DCs,demonstrating enhanced effect of OVA-loaded polymersomes with dual agonists on DCs-based immunotherapy.
OVA-loaded polymersomes were synthesized and characterized in our study.PCL-PEG-PCL polymersomes (PCEP-PS) co-delivery of immunostimulant molecules and OVA modal antigen were fabricated by a W/O/W method.Using this method, PCEP polymer formed an aqueous hydrophilic PEG core to encapsulate OVA inside and hydrophobic PCL membrane layer to incorporated lipophilic TLR7 agonist IMQ.TLR4 agonist MPLA was anchored into the lipid layer and outside OVA was absorbed on the outer layer of the PS surface by electronic interaction with DOTAP lipid (Fig.S1 in Supporting information).The physical parameters of the formulated PCEP-PS and the loading amounts of OVA were summarized in Table S1 (Supporting information).The average size of the PCEP-PS was detected by dynamic light scattering (DLS) and TEM.Both techniques exhibited that the hydrodynamic diameter of these PCEP-PS was ranged from 250 nm to 300 nm with a PDI of 0.31–0.41.Nanoparticles with this size are reported to effectively activate DCs and induce systemic immune responses compared to bigger particles [30].As shown in Fig.S2A (Supporting information), the TEM image showed that OVA-loaded PS had a spherical shape with some visible particles on the surface, demonstrating successfully adsorption of OVA.The zeta potential was slightly negative, indicating that the OVA was successfully absorbed on the outer layerviacationic DOTAP lipid.OVA was efficiently encapsulated in PCEP-PS with a loading amount ranged from 45 μg/mg to 57 μg/mg PS, indicating their great capability of loading antigen(Table S1).
In vitroOVA release study was conducted in mimicking normal physiologic microenvironment (PBS, pH 7.4).Representative cumulative release curve of OVA from polymersomes was shown in Fig.S2B (Supporting information).About 20% of the antigen was released from IMO-PS within 24 h and 70% of the antigen was then continuously released for 28 days.Antigen release is one of the most important factors for particle-based vaccination.Prolonging antigen release can enable antigen persistence to immune cells, which is favorable to induce robust T cell responses [31].Meanwhile, initial antigen exposure is also important due to the quick induction of antigen-specific immune responses.The combined quick and slow antigen release from the well-designed IMOPS provided not only initial antigen exposure but also long-term antigen persistence, which was critical to enhance immune responses.
To ensure the loading of OVA-Cy7 in PECP-PS, we usedin vivoimaging system to acquire NIR fluorescence images of prepared OVA-loaded PS.Fig.S2C (Supporting information) showed that all OVA-loaded PS groups exhibited equivalent fluorescence, while no fluorescence signals were detected in PBS group.To determine the DC labeling efficiency, DCs were incubated with OVA-Cy7 loaded PS at various concentrations (25, 50, 100 or 200 μg/mL) for 16 h and then NIR fluorescence images were obtained.As shown in Fig.S2D (Supporting information), the fluorescence signal intensity increased with the increase of concentration, and intensive NIR fluorescence from the pelleted DCs were observed at concentration higher than 50 μg/mL.
For DC-based immune therapy, the injected DCs should be capable of keeping viable long enough to migrate to the secondary lymphoid organs and trigger an immune response.Thus, the viability of DCs after incubating with OVA-loaded PS was critical to achieve effective therapy.DC viability was assessed after being treated with various OVA-loaded PS at different OVA concentrations (25 and 50 μg/mL) for 24 and 48 h.As shown in Figs.1A and B, no cytotoxic effect was observed on BMDCs after incubating with free OVA or OVA-loaded PS, indicating the high cytocompatibility and safety of the formulated antigen carriers.
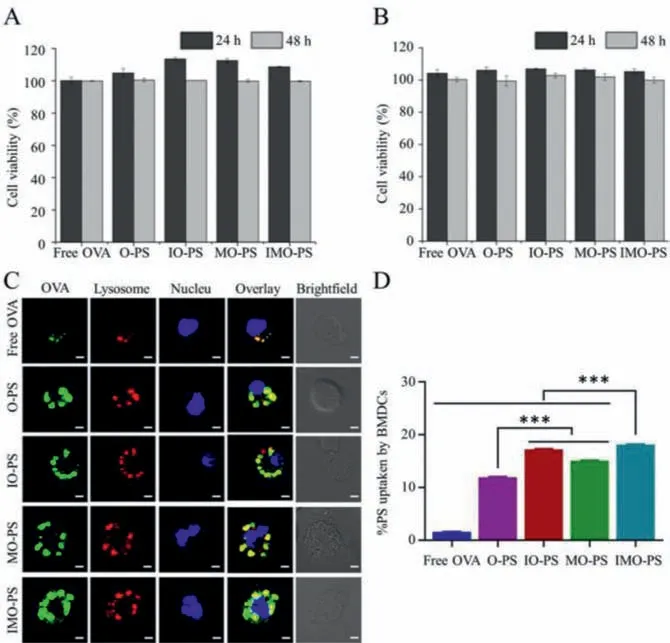
Fig.1.Cell viability assay and in vitro PCEP-PS uptake.(A, B) Viability of DCs after treating with various OVA-loaded PCEP-PS at different OVA concentrations (A:25 μg/mL; B: 50 μg/mL) for 24 and 48 h.(C) CLSM images of antigen intracellular localization after incubating DCs with free OVA, O-PS, IO-PS, MO-PS and IMO-PS for 16 h.(D) The percentage of antigen uptake by DCs using flow cytometry analysis.∗∗∗P < 0.001.
Antigen uptake, the processing and presentation by DCs is an important step for initializing long-time memory and adaptive immunity.CLSM was used to investigate the cellar uptake and lysosome escape.As shown in Fig.1C, obvious green OVA fluorescence was observed in all the PCEP-PS groups, while very weak signal was detected in the free OVA group.This result demonstrated that PCEP-PS greatly facilitated antigen internalization by DCs.In addition, FITC-OVA was perfectly co-localized with lysosomes in free FITC-OVA group, confirming antigen degradation and processingviathe endosome/lysosome pathway.For PCEP-PS groups, although most of FITC-OVA were co-localized with lysosomes, some of them were found in the cytosol.Antigen release into cytosol is beneficial to cross-presentation for the further activation of CD4+and CD8+T cells [32,33].Antigen delivery systems mediated by NPs have been reported to be more efficient in facilitating APCs to crosspresent antigen [34–36].In our study, the lysosome escape might result from the “proton sponge” effect of cationic DOTAP, which could lead to osmotic swelling and lysosome rupture, thereby causing the cytosolic release of antigens [37].The intracellular localization of OVA confirmed that PCEP-PS enabled the endocytic delivery of exogenous antigen for presenting through both MHC I and MHC II pathways.Since TLR7 is primarily localized in the endosomal region of DCs, efficient IMQ released from the membrane of IO-PS and IMO-PS in the endosome could beneficially promote the interaction of TLR7 and IMQ agonist.The effective internalization of IMO-PS by BMDCs could result in the enhanced synergistic immune activation by both TLR4 and TLR7 signaling pathway.
The antigen uptake capability of free OVA and various OVAloaded PS was further quantitatively analyzed by FACS.As shown in Fig.1D, free OVA group showed poor antigen uptake efficacy.Compared to O-PS, the OVA-loaded PS with TLR4 and/or TLR7 agonist (IO-PS, MO-PS and IMO-PS) showed significantly higher OVA delivery efficiency into DCs (Fig.1D).OVA-loaded PCEP-PS remarkably enhanced antigen uptake by DCs than free OVA did, contributing to that the cationic DOTAP on the outer layer of the PCEPPS could significantly facilitate antigen internalization by DCs [38].Compared with O-PS, MO-PS increased antigen uptake by DCs,which is in accordance with the previous study that MPLA incorporated in NPs enhanced uptake efficiency of antigens by APCsviathe interaction of MPLA and extracellular TLR4 agonist.Furthermore, IMO-PS group exhibited a higher cell uptake capability than IO-PS and MO-PS, implying that PS encapsulating dual TLRs could provide more efficient ligand internalization.The results above demonstrated that our polymersomes could act as an effective antigen-delivery system for preparing DCs-based vaccine.
As one of the most important APCs, DCs not only can stimulate innate immune responses but also capture, process and present antigens to naïve T cells to initiate adaptive immune responses.Immature DCs served as antigen-capturing cells, while mature DCs mainly acted as antigen-presenting cells [39].Thus, we analyzed DCs surface markers and secreted cytokines to assess the possible effects of O-PS, IO-PS, MO-PS and IMO-PS on DCs maturation.It was reported that antigen-loaded DCs kept in a resting state in the absence of activation and maturation stimulation, while they could efficiently facilitate adaptive immune responses when being stimulated by danger signals [40].TLR-based immune adjuvants were reported to induce enhanced expression of maturation markers such as costimulatory (CD80, CD86) molecules on DCs [41].The level of CD80 and CD86 was an important index to assess the immune response in DC-based immunotherapy [42].DOTAP liposomes were reported to improve vaccine-induced immune responses by inducing DC maturation [43].In our studies, different OVA-loaded vaccine formulations were cultured with immature DCs for 24 h, and then CD80 and CD86 expression on CD11c+cells were assessed by FACS.As shown in Fig.S3A (Supporting information), IMO-PS significantly induced higher expression of CD80 than free OVA and O-PS group.IO-PS, MO-PS and IMO-PS had higher expression of CD86 than O-PS.More importantly, the MO-PS and IMO-PS with MPLA agonist on the outer layer significantly elevated the expression of CD86 in comparison to other groups.This was probably due to that MO-PS and IMO-PS can mimic bacterial cell walls and enhance phago- and endo-cytosis by DCs as well as increase expression of costimulatory molecules and production of cytokines when MPLA ligating with TLR4 on DCs surface.Our results demonstrated that directly activating TLRs with IMQ and MPLA could synergistically promote DCs maturation.
To assess whether immunostimulatory molecules (IMQ and/or MPLA) in PCEP-PS could affect cytokines secretion, DCs were cultured with O-PS, IO-PS, MO-PS and IMO-PS for 24 h.Cell culture supernatants were harvested and diluted to evaluate cytokines (TNF-α, IFN-γ) secretion using ELISA.As elucidated in Fig.S3C (Supporting information), the secretion of IFN-γfrom BMDCs treated with IO-PS, MO-PS, and IMO-PS was 2.0-, 2.4-, and 10.0-fold higher than that treated by O-PS group.Meanwhile, the generation of TNF-αwas also greatly augmented in PCEP-PS group with IMQ and/or MPLA agonist.As shown in Fig.S3D (Supporting information), IO-PS, MO-PS, and IMO-PS group yielded 5.5-,8.0-, and 9.0-fold higher TNF-αgeneration than O-PS group.More importantly, IMO-PS with dual agonists significantly elevated both IFN-γand TNF-αproduction compared to IO-PS and MO-PS with only single agonist.The activation of TLR7 signaling had the potential to induce DCs maturation and stimulate the secretion of proinflammatory cytokines [44,45].MPLA treated DCs were reported to be more efficient at producing Th1 cell-derived cytokine, IFN-γ[46].Our results demonstrated that the co-delivery of both TLR7 and TLR4 agonist within the polymersomes had synergistic effect on promoting the production of pro-inflammatory cytokines, which were important in DCs-based immunotherapy.
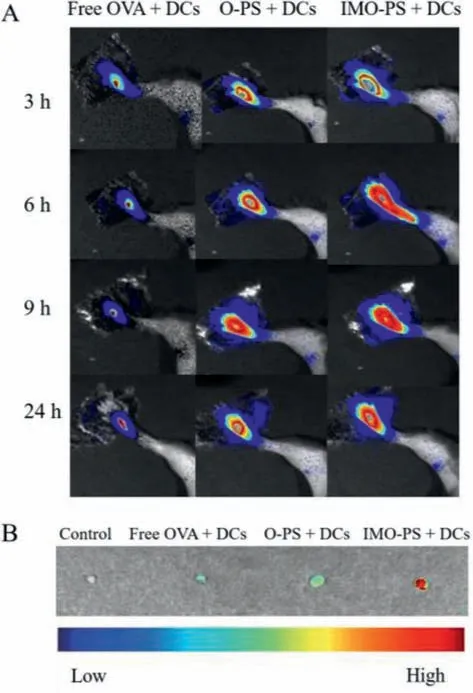
Fig.2. In vivo tracking of various OVA-loaded PS treated DCs (A) NIR images obtained at 3, 6, 9, 24 h after injecting free OVA, O-PS and IMO-PS treated DCs(2×106) into the right footpad of C57BL/6 mice.(B) Inguinal lymph node homing of free OVA, O-PS and IMO-PS treated DCs (PBS treated group was set as control).
Cross-presentation of endocytosed antigens could activate cytotoxic CD8+T cell and then generate antitumor immunity.To evaluate whether the formulated PCEP-PS could enhance antigen MHC I presentation, DCs were first incubated with OVA-loaded PS and then cocultured with OVA-specific LacZ B3Z CD8+T cells hybridoma.B3Z T cells could produceβ-galactosidase during the process of recognizing OVA SIINFEKL, which is presented by MHC I H-2Kb on DCs.And the producedβ-galactosidase was then quantified by CPRG, aβ-galactosidase substrate, to measure antigen crosspresentation.Fig.S3E (Supporting information) showed that compared to free OVA, OVA-loaded PS increased class I antigen presentation which may due to the increased antigen uptake in OVAloaded PS treated DCs.Furthermore, IMO-PS significantly improved the capability of cross-presentation, which may attribute to the combined effect of immune activator IMQ and MPLA.This data indicated that OVA-loaded PCEP-PS with TLR agonist, especially IMOPS with dual agonists, was superior to antigen alone in activating MHC I cross-presentation.
In DC-based immunotherapy, migration of antigen-loaded DCs from administrated sites to draining LNsvialymphatic vessels is essential for the induction of adaptive immune responses [47].After migrating to LNs, DCs present the processed antigens in peptide form to CD4+and CD8+T cells and then initiate proper immunity against the antigens [48,49].Nevertheless, subcutaneously injected DCs were reported to migrate to the draining lymphoid tissue in a minority, while the majority were remained at the injected site [50].Thus, enhancing DCs migration into LNs is a key process to improve DC-based immunotherapeutic effect.To explore whether IMO-PS treated DCs could achieve a higherin vivomigration to the LNs, DCs (2×106cells) incubated with free OVA, OPS, and IMO-PS were injected into a left hind-leg footpad of the C57BL/6 mouse.Then, fluorescence signals were observed by thein vivoimaging system.As shown in Fig.2A, Cy7-OVA fluorescence appeared in the inguinal LNs 3 h post injection of IMO-PS treated DCs and keep strong at 24 h, whereas free OVA and OPS treated DCs showed a relatively weak fluorescence at 3 h post injection and almost no fluorescence at 24 h, implying gradual migration of IMO-PS treated DCs from the administration site to the draining lymph node through lymphatic vessels.Furthermore, DCs migration was confirmed byex vivoNIR images of dissected inguinal LNs.Fig.2B showed that IMO-PS treated DCs generated the strongest NIR signals, thus demonstrating their effective migration to the draining LNs.However, immature DCs treated with free OVA and O-PS showed a weak signal.The capability of DCs to migrate into draining LNs is essential for DCs to exert functions.Our results demonstrated that IMO-PS treated DCs remained active and achieved migration to LNs after being injected back into mice.
To confirm antigen-specific T cell responses in immunized mice,intracellular IFN-γwas evaluated by FACS after 96 hex vivostimulation with SIINFEKL peptide.Co-delivery of antigen and adjuvant to DCs was reported to be important to induce strong CD8+T cell immune response [51].TLR activation of DCs is capable of priming antigen-specific CD4+T-helper cells, which are essential to induce antibody responses [52,53].Representative FACS plots and mean frequencies of IFN-γproducing CD4+and CD8+T cells were shown in Fig.3.As for IFN-γ+CD4+T cell in spleen, the percentage in mice immunized with IMO-PS, IO-PS and MO-PS treated DCs was 1.68% ± 0.12%, 1.22% ± 0.14%, and 1.33% ± 0.10%, respectively.On the contrary, PBS, free OVA and O-PS treated DCs immunized mice were significantly lower, with a percentage of 0.52% ± 0.12%,0.53% ± 0.14%, and 0.55% ± 0.11%, respectively.The percentage of IFN-γ+CD8+T cells detected in spleen of mice immunized with IMO-PS treated DCs was 2.44% ± 0.11%, which was significantly higher than IO-PS treated DCs (1.25% ± 0.13%) and MO-PS treated DCs (1.29% ± 0.11%).Additionally, the amount of IFN-γ+CD8+T cells was remarkably lower in mice immunized with PBS, free OVA and O-PS treated DCs, with a percentage of 0.68% ± 0.12%,0.69% ± 0.14% and 0.74% ± 0.11%, respectively.The data suggested that IMO-PS greatly improved the ability of DCs to induce antigenspecific CD8+and CD4+T cell responses.
The OVA-loaded polymersomes could also induce OVA-specific CTL responsein vivo.CTLs, a type of T lymphocytes, can be activated by mature DCs [54] and kill infected cells or cancer cells by releasing perforin and granzymes [55].Antigen-loaded DC-based vaccines have been considered to directly induce a specific CTL response [56].Considering the increased MHC I presentation and IFN-γproduction by IMO-PS, we speculated that IMO-PS would effectively stimulate vaccine-induced CTL responses.To evaluate the OVA-specific CTL response induced by PS, mice were immunized with various PCEP-PS treated DCs.7 days after administration, splenocytes were obtained and re-stimulated with OVA peptide (SIINFEKL) in 1640 medium containing IL-2 for 72 h.Calcein-AM release assay was then used to evaluate cell viability and assess the CTL activity.The results showed that splenocytes harvested from mice immunized using IMO-PS treated DCs effectively induced CD8+T-mediated tumor cell lysis (Fig.S4A in Supporting information).The enhanced CTL responses could be contributed to the increasement of MHC I presentation and IFN-γproduction by IMO-PS.On the contrary, immunization with free OVA and OPS treated DCs failed to stimulate effective tumor specific CTL responses, in accordance with their poor effect on MHC I presentation and IFN-γsecretionin vitro.Compared with free OVA and OPS, the greatest cytotoxicity against EG7-OVA was achieved at the E:T ratio of 40:1 (Fig.S4B in Supporting information).The above results implied that IMO-PS treated DCs induced efficient antigenspecific CTL activity, demonstrating the synergistic effect of TLR4 and TLR7 signaling pathway on cellular immunity.
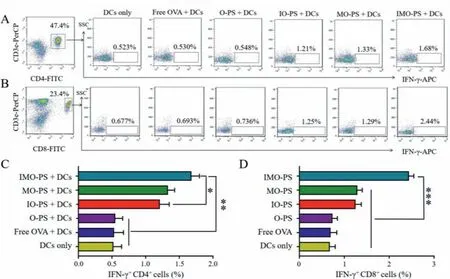
Fig.3.Intracellular IFN-γ production was analyzed by FACS after splenocytes were restimulated with OVA peptide (1 μmol/L) for 96 h (n=3).Representative FACS plots of (A) IFN-γ producing CD4+ T cells and (B) IFN-γ producing CD8+ T cells.Mean frequencies of (C) IFN-γ producing CD4+ T cells and (D) IFN-γ producing CD8+ T cells.∗P < 0.05, ∗∗P < 0.01 and ∗∗∗P < 0.001.
To investigate the protective efficacy of PCEP-PS treated DCs against progressive EG7-OVA tumor, we immunized C57BL/6 mice with free OVA, O-PS, IO-PS, MO-PS, IMO-PS treated DCs (Fig.4A).PBS treated DCs were set as control group.One week after immunization, 1×106EG7-OVA cells were inoculated subcutaneously on the right back and the percentage of tumor-free mice was evaluated (Fig.4B).All mice in PBS+DCs group developed tumors within 4 days, while the mice injected with free OVA or O-PS treated DCs delayed to develop tumors within 12 days after tumor cell injection.On the contrary, 25% of mice administrated with IO-PS+DCs and MO-PS+DCs remained tumor free on day 54 post-challenge.Strikingly, the tumor free percentage was further increased to 37.5% for the mice vaccinated with IMOPS+DCs (Fig.4B).The survival rate was calculated up to 54 days after tumor inoculation.The mice vaccinated with IMO-PS showed 37.5% survival rate, while the IO-PS and MO-PS treated mice had 25% survival rate (Fig.4C).The results above suggest that IMOPS can elicit potent therapeutic anti-cancer immunity.Remarkably,the tumor-free mice in the IO-PS+DCs, MO-PS+DCs and IMOPS+DCs treated groups were re-challenged with 1×106EG7-OVA tumor cells on day 95 and they remained 100% tumor-free on day 155 (Fig.4D), confirming their capability of exhibiting long-term immunity against tumor reoccurrence.Combination of TLR4 and TLR7 ligands enhanced immune responses by synergistic effectviatriggering different pathways and thus influenced antigen dependent T-cell immune memory.These data above demonstrated that the combinational delivery of OVA, imiquimod and MPLA synergistically triggered anti-tumor immune responsesin vivo, thus achieving superior preventive effect against tumor challenge.
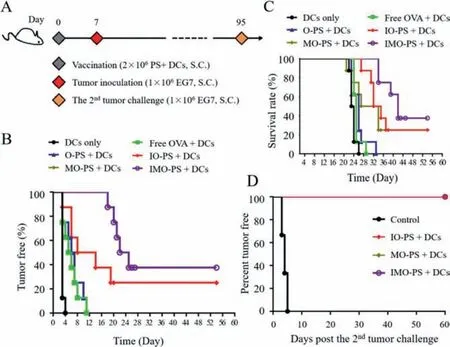
Fig.4. In vivo protective efficacy of PS treated DCs.(A) Immunization schedule.(B)Percentage of tumor free mice after EG7-OVA tumor cells challenge.(C) Survival rate of EG7-OVA tumor-bearing mice.(D) Re-challenge study on tumor free mice in the IO-PS+DCs, MO-PS+DCs and IMO-PS+DCs group.Naïve mice (n=6) were used as the control group.
In summary, we have constructed cationic hybrid polymersomes (IMO-PS) for programmed co-delivery of OVA antigen and dual-adjuvants (IMQ and MPLA) for DC-based immunotherapy against cancer.The IMO-PS dramatically enhanced the OVA uptake in DCs and promoted DCs maturation and cytokines production.In addition, IMO-PS+DCs greatly improved DCs homing ability and OVA-specific CTL responses.Moreover, IMO-PS+DCs significantly extended tumor-free time, effectively suppressed tumor growth and greatly extended median survival time on the prophylactic EG7-OVA tumor model.Impressively, the tumor-free mice were re-challenged with EG7-OVA tumor cells on day 95 and they remained 100% tumor-free at the end of the observation time (155 days).The formulated co-delivery nanoplatform with dual agonists effectively stimulates DCs and holds great promise to be developed as DC-based vaccine for future immunotherapy treatment against cancer.
Ethical compliance
All animals were treated according to the regulations and guidelines of the Institute of Biomedical Engineering, Chinese Academy of Medical Science & Peking Union Medical College.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was financially supported by National Natural Science Foundation of China (Nos.82072059 and 82172090),CAMS Initiative for Innovative Medicine (No.2021-I2M-1-058), the Fundamental Research Funds for the Central Universities (Nos.2019PT320028 and 2019-0831-03), Tianjin Municipal Natural Science Foundation (No.20JCYBJC00030).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2022.02.022.
 Chinese Chemical Letters2022年9期
Chinese Chemical Letters2022年9期
- Chinese Chemical Letters的其它文章
- A review on recent advances in hydrogen peroxide electrochemical sensors for applications in cell detection
- Rational design of nanocarriers for mitochondria-targeted drug delivery
- Emerging landscapes of nanosystems based on pre-metastatic microenvironment for cancer theranostics
- Radiotherapy assisted with biomaterials to trigger antitumor immunity
- Development of environment-insensitive and highly emissive BODIPYs via installation of N,N’-dialkylsubstituted amide at meso position
- The effect of organic ligand modification on protein corona formation of nanoscale metal organic frameworks