
Nickel-catalyzed reductive cross-coupling of polyfluoroarenes with alkyl electrophiles by site-selective C–F bond activation
2022-09-15 03:11LonglongXiLitingDuZhungzhiShi
Chinese Chemical Letters 2022年9期
Longlong Xi, Liting Du, Zhungzhi Shi,∗
a State Key Laboratory of Coordination Chemistry, Chemistry and Biomedicine Innovation Center (ChemBIC), School of Chemistry and Chemical Engineering,Nanjing University, Nanjing 210093, China
b Advanced Analysis and Testing Center, Nanjing Forestry University, Nanjing 210037, China
ABSTRACT A nickel-catalyzed reductive cross-coupling reactions between polyfluoroarenes and alkyl electrophiles is reported to access substituted fluoroarenes through chelation-assisted C–F activation.Diverse primary and secondary alkyl (pseudo)halides can be employed to couple with polyfluoroarenes, showing excellent regioselectivity.Furthermore, the nickel-catalyzed asymmetric cross-coupling of polyfluoroarenes with racemic alkyl halides is preliminarily explored.In addition, the practicability of the title transformation is also demonstrated by total synthesis of losmapimod and an analog as key steps.The developed method exhibits many advantages, including economic catalytic systems, commercially available alkyl el ectrophiles, and lack of sensitive organometallic reagents.
Keywords:Polyfluoroarenes Nickel Alkylation Cross-coupling C-F activation
Aryl fluoride is among the most versatile structural motifs in pharmaceuticals and biologically active molecules [1–5].During the past decades, extensive efforts have been made by chemists toward the exploration of more general and practical methods to build them.Compared to the common fluorination process [6–12],defluorinative functionalization of polyfluoroarenes has emerged as a promising way to generate unique fluorinated compounds[13–38].An illustrative case is described in Fig.1a, wherein losmapimod (I), as a selective MAPK (mitogen-activated protein kinases) inhibitor, was accessed by de novo synthesis from compound II through a palladium-catalyzed Suzuki-Miyaura coupling reaction [39].We envisioned that late-stage defluoroalkylation of a difluoroarene III might provide an alternative route to molecule I, especially simplifying the route to a series of analogs bearing diverse alkyl substituents, for high-throughput screening in drug discovery.
Typically, preformed alkyl organometallic reagents (Zn, Mg, Li)need to be employed in the defluoroalkylation of polyfluoroarenes,leading to inconvenient operation and limited substrate scope [40–43].Recent progress has provided new defluoroalkylative methods from sustainable chemical feedstocks [44,45].For example,Xionget al.developed Cu-catalyzed defluoroalkylation of polyfluoroarenes using alkenes and silanes (Fig.1b, left) [46].The Ritter group also uncovered the related defluorinative alkylation by decarboxylation of aliphatic carboxylic acids under photoredox conditions (Fig.1b, right) [47].Controlled by electronic and steric effects,such reactions exhibit good para-selectivity in symmetric polyfluoroarenes, but the use of unsymmetrical substrates leads to low regioselectivity.
Transition metal-catalyzed reductive cross-coupling reactions,which employ two electrophiles with a terminal reductant,have emerged as inspiring methods for the construction of carbon−carbon bonds [48–55].Aryl [56–64] or vinyl [65–67]halides, including Cl, Br, I and pseudohalides such as OTs and OTf,have been used for the synthesis of alkylated arenes with alkyl electrophiles in the presence of nickel catalysis (Fig.1c) [68–74].Given the high value of fluorinated arenes, the direct use of polyfluoroarenes in reductive cross-coupling through C–F activation is appealing [75–77].Here, we showcase the first example of site-selective defluoroalkylation of polyfluoroarenes with benchstable and readily available alkyl (pseudo)halides, avoiding the pregeneration of air- and moisture-sensitive organometallic reagents(Fig.1d).Controlled by the chelation effect, diverse polyfluoroarenes with coordinating functional groups direct nickel to activate proximal C–F bondsviacyclometallated intermediates, showing an excellent level of ortho-selectivity [78–83].
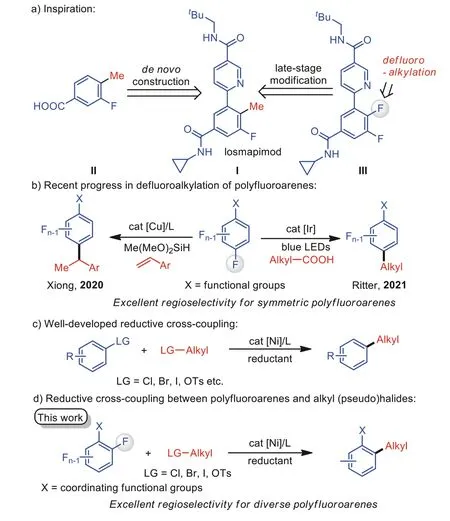
Fig.1.Nickel-catalyzed defluoroalkylation of polyfluoroarenes with alkyl electrophiles through reductive cross-coupling reactions.
We began our work by monitoring the reactivity of polyfluoroarene 1a and alkyl bromide 2a (Table 1).After systematic screening, the reaction was found to be facile with NiBr2(dme) (10 mol%),X-phos (12 mol%), Zn0(2.0 equiv.) as the stoichiometric reductant, NaI (2.0 equiv.) as the additive in DMA after 2 h at 50°C,affording desired defluoroalkylation product 3aa in 80% yield (entry 1).Diphosphine ligands such as xantphos (L2) greatly lowered the selectivity (entry 2).Several bidentate nitrogen ligands were also investigated;tert-butyl bipyridine L3 delivered product 3aa in 60% yield (entry 3), and a bis(oxazoline) ligand L4 could improve the yield to 71% (entry 4).The use of Zn0can accelerate the cross-coupling process, and other reductants, such as Mn0, were not effective (entry 5).Notably, shortening reaction time to 45 min maintained a good reactivity (entry 6), and extending the reaction time to 12 h led to the slow decomposition of product 3aa (entry 7).In addition, conducting the reaction at room temperature resulted in a slightly lower yield (entry 8).The reaction completely failed in the absence of NaI, which was reported to be a key additive in reductive cross-couplingsviaacceleration of electron transfer between metal reductant and nickel catalyst (entry 9) [84–87].In addition, treatment of alkyl iodide 2a’in the reaction showed slightly higher reactivity, affording the product 3aa in 82% yield(entry 10).Under the above reaction conditions, removal of NaI in the system could detect the formation of the product 3aa, albeit with low conversion, indicating that NaI may also help in situ formation of organo-iodide electrophiles from alkyl bromides in the system (entry 11).Finally, a control experiment confirmed that the defluoroalkylation failed without the addition of Ni catalyst (entry 12).
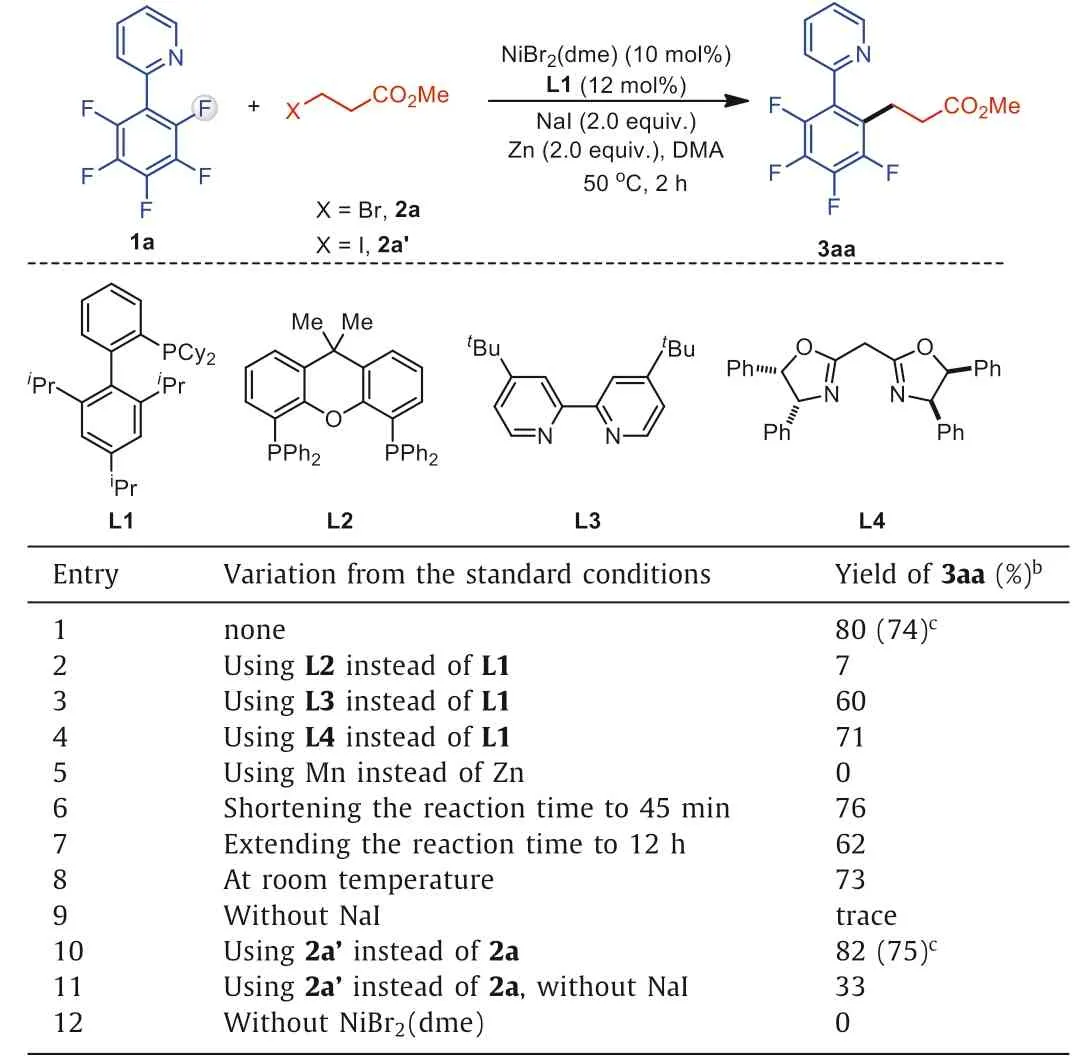
Table 1 Reaction optimization.a
With the optimized reaction conditions in hand, we first examined the scope of alkyl electrophiles (Scheme 1).A range of primary alkyl bromides and iodides were efficiently transformed into the corresponding products, including those possessing ester(3ab and 3ac), aryl (3ad and 3ae), and fluoroalkyl (3af-3ai).Among these compounds, the structure of 3ae was further confirmed by Xray diffraction.Moreover, selective defluoromethylation of polyfluoroarene 1a using CH3I (2j) provided product 3aj in 51% yield [88].Alkyl tosylate 2l and secondary alkyl iodide 2m were also tolerated, in which the selection of ligand L4 exhibited much higher reactivity.Under slightly modified reaction conditions, cyclic compounds 2n and 2o underwent facile coupling.Furthermore, a vari-ety of benzyl chlorides 2p-2s andβ-substituted derivatives 2t and 2u could also maintain good reactivity.We were pleased to find that alkyl electrophiles 2v and 2w (I, Br, OTs) bearing a remote aryl group were also compatible with this reaction, and a reported chain walking process to the benzylic position was inhibited in our system [89,90].
We next subjected various polyfluoroarenes to the system with alkyl bromide 2a (Scheme 2).A broad scope of polyfluoroarenes bearing four (1b and 1c), three (1d-1f), and two (1g) fluorine atoms underwent cross-coupling to furnish products 3ba-3ga in 22%-94% yields with excellent regioselectivities.It’s found that the lower the number of substituted fluorine atoms was, the lower the yield of products, and monofluoroarene 1h gave only trace amounts of product 3ha.A series of polyfluoroarenes with substituents including Me (1i), OMe (1j and 1k), NMe2(1l), CF3(1n) and Ph (1o and 1p) moieties were competent coupling partners.The reductive cross-coupling can be extended to heteroaromatic systems;e.g.,polyfluoropyridine 1q was converted to 3qa in 59% yield.Other heteroarenes, such as pyrazine (1r) and benzo[d]oxazole (1s) could also be utilized as directing groups.In addition, the reaction of substrate 1t containing a removable imine-directing group [40,41], can selectively activate two C–F bonds, affording dual alkylation product 3ta in 66% yield.
Our preliminary screening demonstrated the feasibility of the enantioselective reductive cross-coupling of polyfluoroarenes using a chiral nickel catalyst (Scheme 3).Thus, asymmetric induction in the reaction of polyfluoroarene 1a and benzyl chloride 2p was observed using a host of chiral bis(oxazoline) ligands having different backbones, among which (4S,5R)-L4 displayed the highest enantiomeric ratio (er) value of 73:28 with 75% yield.Under the current reaction conditions, a variety of benzyl chlorides including 2q, 2r and 2t could be coupled with 1a to afford chiral products 3aq, 3ar and 3at in moderate to good er values.While further screening of reaction system is currently underway to improve the enantioselectivity, these results would serve as a testament to the reaction mechanism involving an achiral benzyl radical during the coupling process [91–93].
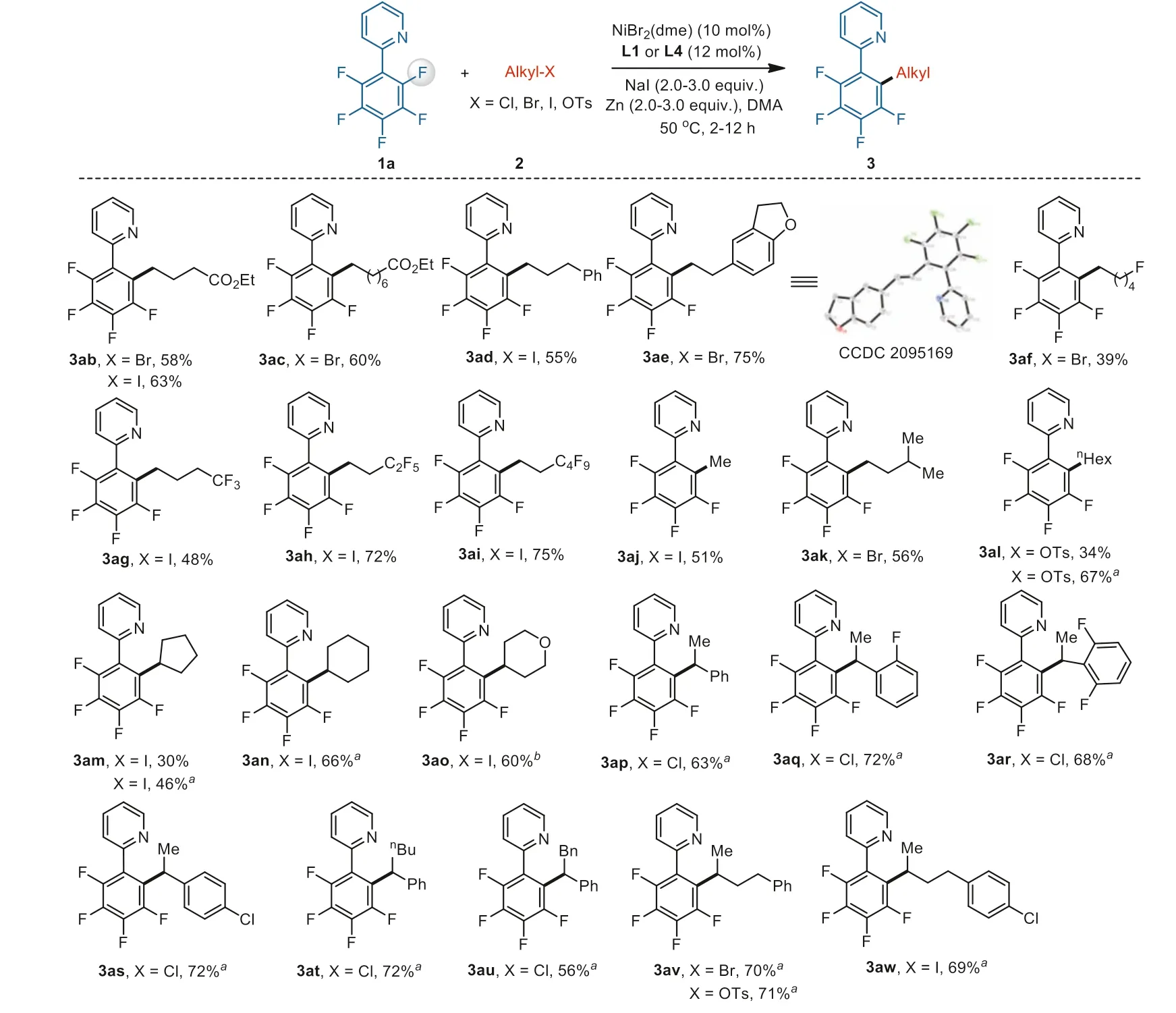
Scheme 1.Substrate scope of alkyl electrophiles.Reaction conditions: 10 mol% NiBr2(dme), 12 mol% L1, 1a (0.20 mmol), 2 (0.40 mmol), NaI (0.40 mmol), Zn (0.40 mmol),DMA (1 mL), 50°C, 2 h; isolated yields.a12 mol% L4, 2 (0.60 mmol), NaI (0.60 mmol), Zn (0.60 mmol), 12 h.
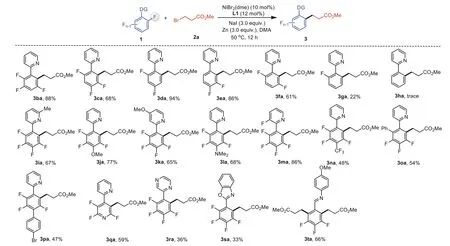
Scheme 2.Substrate scope of polyfluoroarenes.Reaction conditions: 10 mol% NiBr2(dme), 12 mol% L1, 1a (0.20 mmol), 2 (0.60 mmol), NaI (0.60 mmol), Zn (0.60 mmol),DMA (1 mL), 50°C, 2 h; isolated yields.
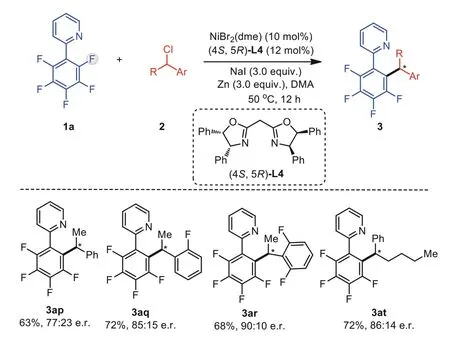
Scheme 3.Preliminary investigation of enantioselective variants.

Scheme 4.Gram-scale transformation and successive defluoroalkylation of polyfluoroarene 1a.
Further investigations were conducted to show the practical utility of the defluoroalkylation process (Scheme 4).The developed system enabled the gram-scale synthesis of 3aa (1.36 g, 87%)from substrates 1a and 2a with higher reactivity compared to the small-scale reaction.Successive defluoroalkylation at different C–F bonds with predictable site selectivity can offer an opportunity to produce complex molecules.For example, polyfluoroarene la has been shown to undergo site-selective defluoroalkylation at thepara-position with styrene 4 to build product 5 in an excellent yield [46].Under our reaction conditions, further ortho-selective defluoroalkylation of compound 5 with alkyl bromide 2a afforded product 6 in 91% yield.
In order to prove the feasibility of the aforementioned assumption in Fig.1a, we next tested the developed method as a key step in the total synthesis of losmapimod (I) (Scheme 5).Our route started with difluoroarene III obtained through Pd-catalyzed Suzuki-Miyaura coupling [39].Under the developed reaction conditions, late-stage and site-selective defluoromethylation of compound III with CH3I (2j) gave product I in 24% yield.Indeed, compared to the method ofde novosynthesis, such a strategy can greatly simplify the route to a library of losmapimod analogs for drug discovery.Using alkyl bromide 2a as a model substrate, latestage defluoroalkylation of III afforded a losmapimod analog 7 in 77% yield.
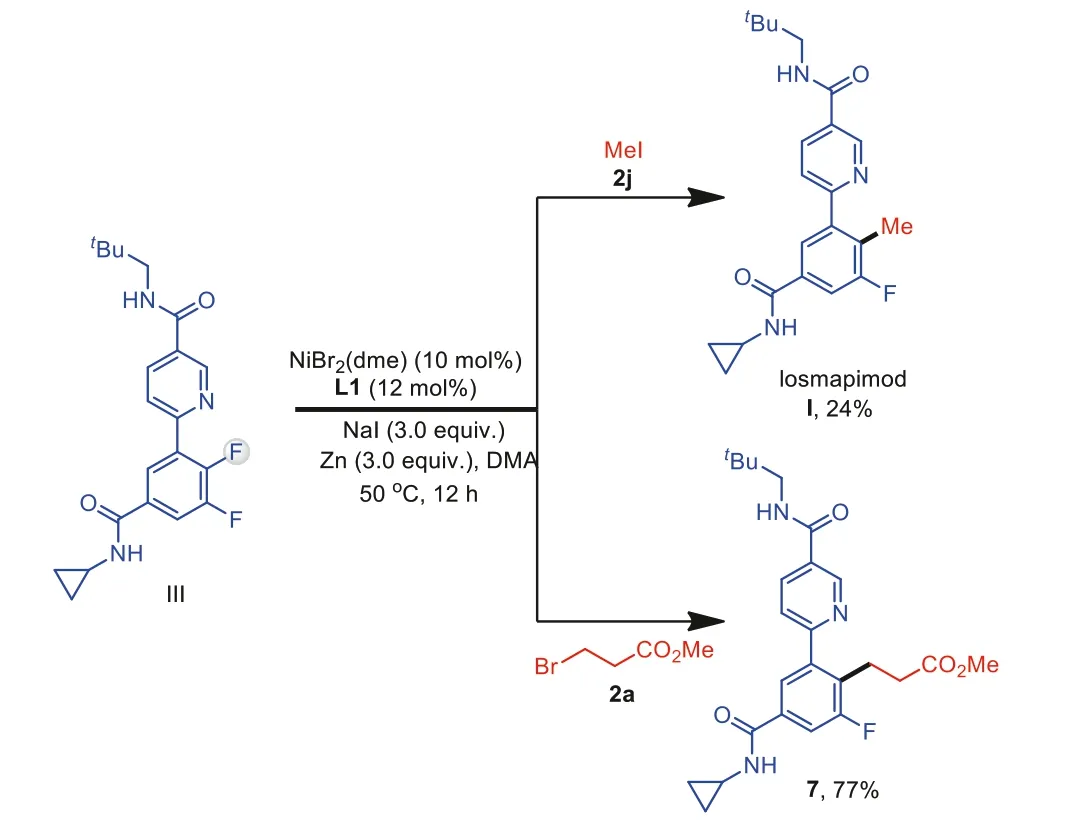
Scheme 5.Synthesis of losmapimod (I) and an analog 7 by late-stage defluoroalkylation of difluoroarene III.
To establish the mechanism for this transformation, we performed several additional experiments (Scheme 6).When a stoichiometric amount of TEMPO was added to the mixture of substrates 1a and 2b, the reaction was completely suppressed (Scheme 6a).Moreover, the addition of 1,1-diphenylethylene to the reaction led to a lower yield of 3ab, and adducts 8 and 9 were detected by GC-MS.These findings suggest the formation of alkyl radicals during cross-coupling.Furthermore, subjecting enantioenriched benzyl halide 2p to the optimized reaction conditions with polyfluoroarene la only afforded racemic product 3ap, demonstrating that an achiral benzyl radical was formed in the reaction (Scheme 6b)[94].When polyfluoroarene 10 bearing two pyridyl groups was allowed to react with alkyl bromide 2a under the standard reaction conditions, the corresponding product 11 was obtained in 27%yield (Scheme 6c).The reaction of substrate 10 with Ni(cod)2at 50°C in THF generated a cyclometalated NiIIcomplex 12 through oxidative addition of Ni0to the C–F bond.Subjecting complex 12 to alkyl halide 2a under the developed system also afforded the target molecule 11 in 47% yield.These results indicate that complex 12 is a viable intermediate in the catalytic cycle.Finally, dissolving complex 12 in chloroform formed a NiII-Cl complex 13, the structure of which was also confirmed by X-ray crystallographic analysis.
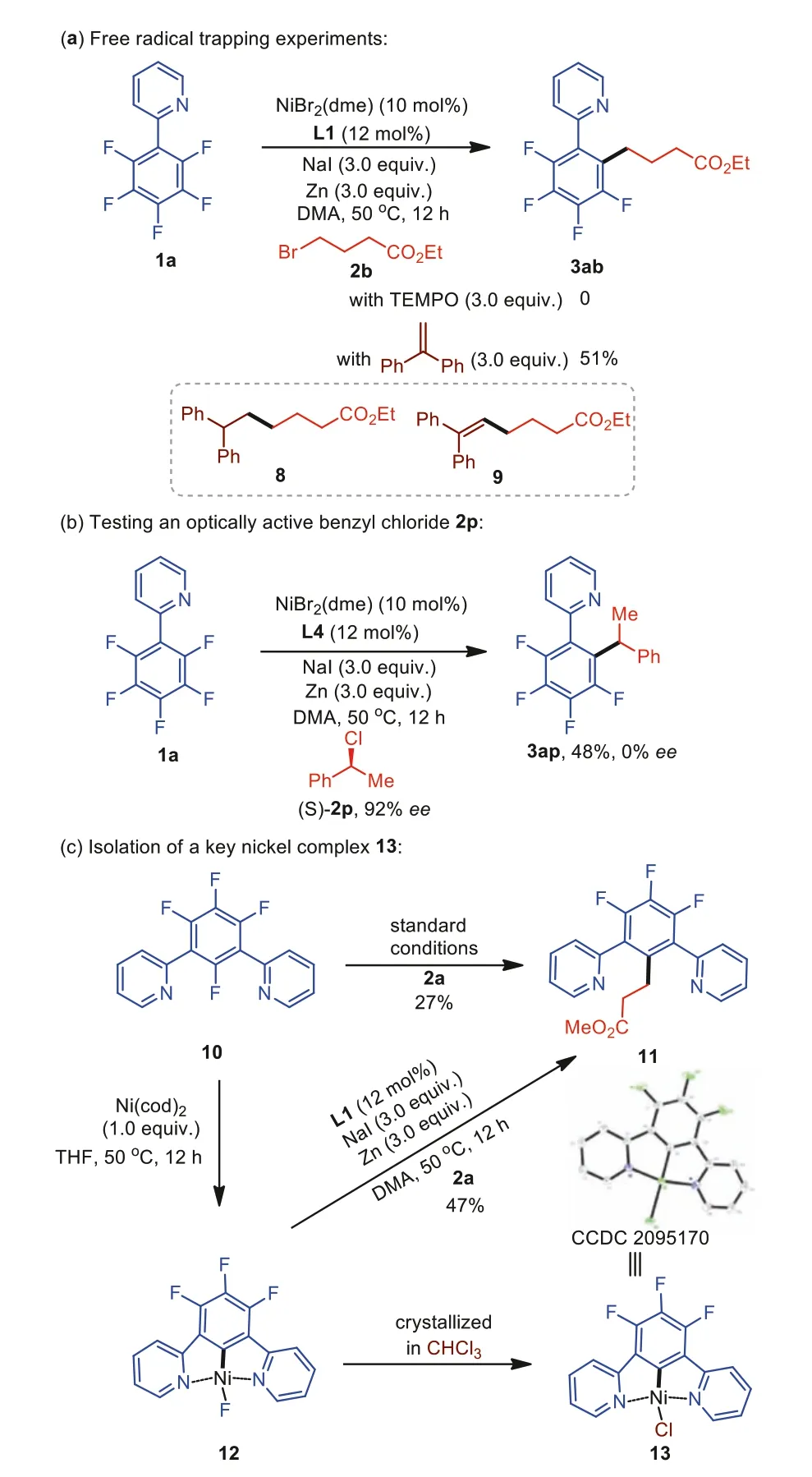
Scheme 6.Mechanistic experiments.
Based on the above results and those in previous works [95–98], a possible reaction mechanism is proposed (Fig.2).Ni0species A undergoes oxidative addition to the C–F bond of polyfluoroarene 1 chelation-assisted by directing groups, affording NiIIcomplex B,which is further reduced by Zn0to generate NiIintermediate C.Alkyl electrophile 2 reacts with Cviaa single electron transfer pathway to form NiIIcomplex D, which further combines with cage-escaped alkyl radicals to generate NiIIIspecies E.Subsequent reductive elimination of E delivers final product 3 and generates NiIspecies F.This species can be further reduced by Zn0, thus forming Ni0species to complete the catalytic cycle.
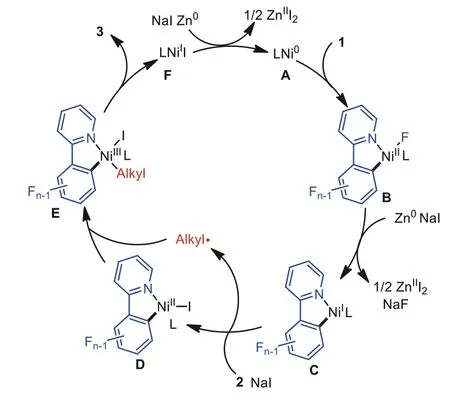
Fig.2.Proposed mechanism.
In summary, we have developed the first defluoroalkylation of polyfluoroarenes with alkyl electrophiles through Ni-catalyzed reductive cross-coupling reactions.Because the coordinating functional groups assist the oxidative addition of the C–F bond, these reactions occur with high reactivity and site selectivity.Further exploration of the enantioselective variant of this defluoroalkylation and examination of detailed mechanistic features are currently ongoing and will be reported in due course.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
We would like to thank the National Natural Science Foundation of China (Nos.22025104, 21972064 and 21901111), the National Natural Science Foundation of Jiangsu Province (No.BK20170632), the Excellent Youth Foundation of Jiangsu Scientific Committee (No.BK20180007), the “Innovation & Entrepreneurship Talents Plan” of Jiangsu Province and the Fundamental Research Funds for the Central Universities for their financial support.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2022.01.077.
 Chinese Chemical Letters2022年9期
Chinese Chemical Letters2022年9期
- Chinese Chemical Letters的其它文章
- A review on recent advances in hydrogen peroxide electrochemical sensors for applications in cell detection
- Rational design of nanocarriers for mitochondria-targeted drug delivery
- Emerging landscapes of nanosystems based on pre-metastatic microenvironment for cancer theranostics
- Radiotherapy assisted with biomaterials to trigger antitumor immunity
- Development of environment-insensitive and highly emissive BODIPYs via installation of N,N’-dialkylsubstituted amide at meso position
- Programmed polymersomes with spatio-temporal delivery of antigen and dual-adjuvants for efficient dendritic cells-based cancer immunotherapy