
A general strategy for in situ assembly of light-up fluorophores via bioorthogonal Suzuki-Miyaura cross-coupling
2022-09-15 03:10XiangLiHongYangYuTengYongchengWangDaliYinYulinTian
Chinese Chemical Letters 2022年9期
Xiang Li, Hong Yang, Yu Teng, Yongcheng Wang, Dali Yin, Yulin Tian
Key Laboratory of Bioactive Substances and Function of Natural Medicine, Beijing Key Laboratory of Active Substances Discovery and Drugability Evaluation,Institute of Materia Medica, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100050, China
ABSTRACT Herein we presented a general strategy for in situ assembly of intramolecular charge-transfer (ICT)-based light-up fluorophores via bioorthogonal Suzuki-Miyaura cross-coupling reaction.By introducing iodo group at the appropriate position, five fluorophores with different scaffolds including naphthalimide,coumarin, naphthalene sulfonate, nitrobenzoxadiazole, and acetonaphthone, were designed as bioorthogonal multicolor fluorogenic probes, which could produce significant fluorescence enhancement and high fluorescence quantum yield after Suzuki-Miyaura reaction with aryl boronic acid or boronate.Manipulating the substituents and π scaffold in the fluorophores allows fine-tuning of their photophysical properties.With this strategy, we succeeded in peptide conjugation, no-wash fluorogenic protein labeling, and mitochondria-selective bioorthogonal imaging in live cells.
Keywords:Bioorthogonal reaction Suzuki-Miyaura cross-coupling Fluorogenic probes Naphthalimide Live-cell imaging
Small-molecule based fluorescence imaging techniques are vital tools for interrogating biological system due to their high sensitivity and fast response time, which have facilitated our understanding of cellular function and biological processes [1,2].However,applications of small-molecule dyes often suffer from unwanted background signals resulting from the unreacted fluorophores and unspecific localization.An alternative approach to overcome these problems is to use fluorogenic probes that possess quenched fluorescence until a specific reaction with their targets [3,4].Compared with the “always-on” type fluorescent probes, these “light-up” type probes have great signal-to-noise ratio that avoid extensive washing cycles to eliminate the background fluorescence.
In recent year, bioorthogonal activated fluorogenic probes have been at the center of attention due to their excellent performance in the bioimaging application, which are designed based on the bioorthogonal reaction with decent kinetics and superior specificity in physiological environments [5].These probes are equipped with a bioorthogonal reporter which is also a fluorescence quencher, and the quenching ability can be abolished upon bioorthogonal reaction, which initiates the “light-up” fluorescence[6].To date, this advanced concept was successfully utilized for the development of numerous fluorogenic probes, where the fluorescence is quenched by azido [7,8], phosphine [9], sydnone [10–12],nitroso [13,14], nitrile oxide [15], tetrazine [16–20],etc.In consideration of the complexity of biological system, there is still a high demand in alternative molecular design strategies for new bioorthogonal applicable light-up probes.
Suzuki-Miyaura cross-coupling, known as the indispensably method for formation of C–C bond in modern organic synthesis, has recently emerged as an attractive bioorthogonal reaction[21].In pioneering work, the Davis group has developed an effi-cient Palladium-mediated Suzuki-Miyaura cross-coupling catalytic system for protein conjugation and cell-surface labeling [22–24].The boronic acid and halogen atom (bromine or iodine) could be treated as the smallest bioorthogonal pair, which has minimal interference in the molecular interactions.On the other hand, as the electron-withdrawing group, halo group is the perfect fluorescence quencher for intramolecular charge-transfer (ICT)-based fluorophore, which usually have donor-π-acceptor (D-π-A) system[25].We envisioned that replacement of the electron donor with halo group could convert the D-π-A system (“push-pull”) to A-π-A system (“pull-pull”), thus block the ICT process, resulting in fluorescence quenching.After Suzuki-Miyaura coupling reaction, new C–C bond was formed and electron-donating aromatic ring was conjugated to the fluorophore as the electron donor, which rebuilt the “push-pull” system and restore the ICT process, producing enhanced fluorescence (Scheme 1).In this contribution, we designed and synthesized five iodo-modified ICT-based fluorophores with different scaffolds.Their fluorescence could be triggered by Suzuki-Miyaura reaction with aryl boronic acid or boronate, giving rise to thein situassembly of light-up fluorophores.This strategy has been successfully applied for fluorogenic labeling of boronic acid/iodo-modified peptide and protein.More importantly, these probes were found to be highly specific and selective for visualizing subcellular organelle such as mitochondria in live cells under no-wash condition.
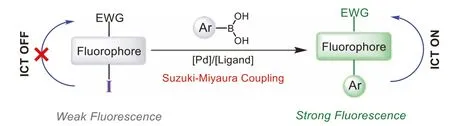
Scheme 1.Design of iodo-modified fluorophores as bioorthogonal light-up probes.
We initially aimed at 1,8-naphthalimide, the most common ICT fluorophore which has immense potential in the area of optical devices, fluorescent sensors, and bioimaging agents [26].The most active halo group in the Suzuki-Miyaura reaction, iodo,was installed on 4-position of 1,8-naphthalimide (NP-I, Table 1)viaSandmeyer reaction (Supporting information).To our satisfaction, the weak visual fluorescence (Figs.1a-c) and low fluorescence quantum yield (ΦF=0.006) verified that the iodo group acts as a good quencher for naphthalimide fluorophore.Then Suzuki-Miyaura coupling was performed between NP-I and phenylboronic acid (1) in 0.1 mol/L Na2HPO4buffer (pH 7.4) at 37°C in the presence of Pd(OAc)2(ADHP)2, a catalytic system developed by the Davis group [22].To our delight, fluorescent 4-phenyl-1,8-naphthalimide (NP-Sz1) was obtained in 34% yield(Table 1, entry 1).Addition of phosphine ligand such as 3-(di–tertbutylphosphonium)propane sulfonate (DTBPPS) or sodium 3,3′,3′′-phosphinetriyltribenzenesulfonate (TPPTS) could significantly improve the reaction efficiency.We found that Pd(OAc)2(5%)/TPPTS(5%) system was preferable to catalyze the reaction in high yield(89%) (Table 1, entry 3).The second-order rate constant (k2) under this condition was calculated to be 1.5 L mol−1s−1(Fig.S1 in Supporting information), which is comparable with those of other bioorthogonal reactions [27].Increasing the amounts of boron reagent or Pd(OAc)2/TPPTS could not significantly improve the yield, while reducing the catalyst loadings decreases the yield obviously (Table 1, entries 4–6).Moreover, similar reaction yield(87%) was observed for the reaction between NP-I and phenylboronic acid pinacol ester (Table 1, entry 7), confirming the same reaction activity between boronic acid and boronate.
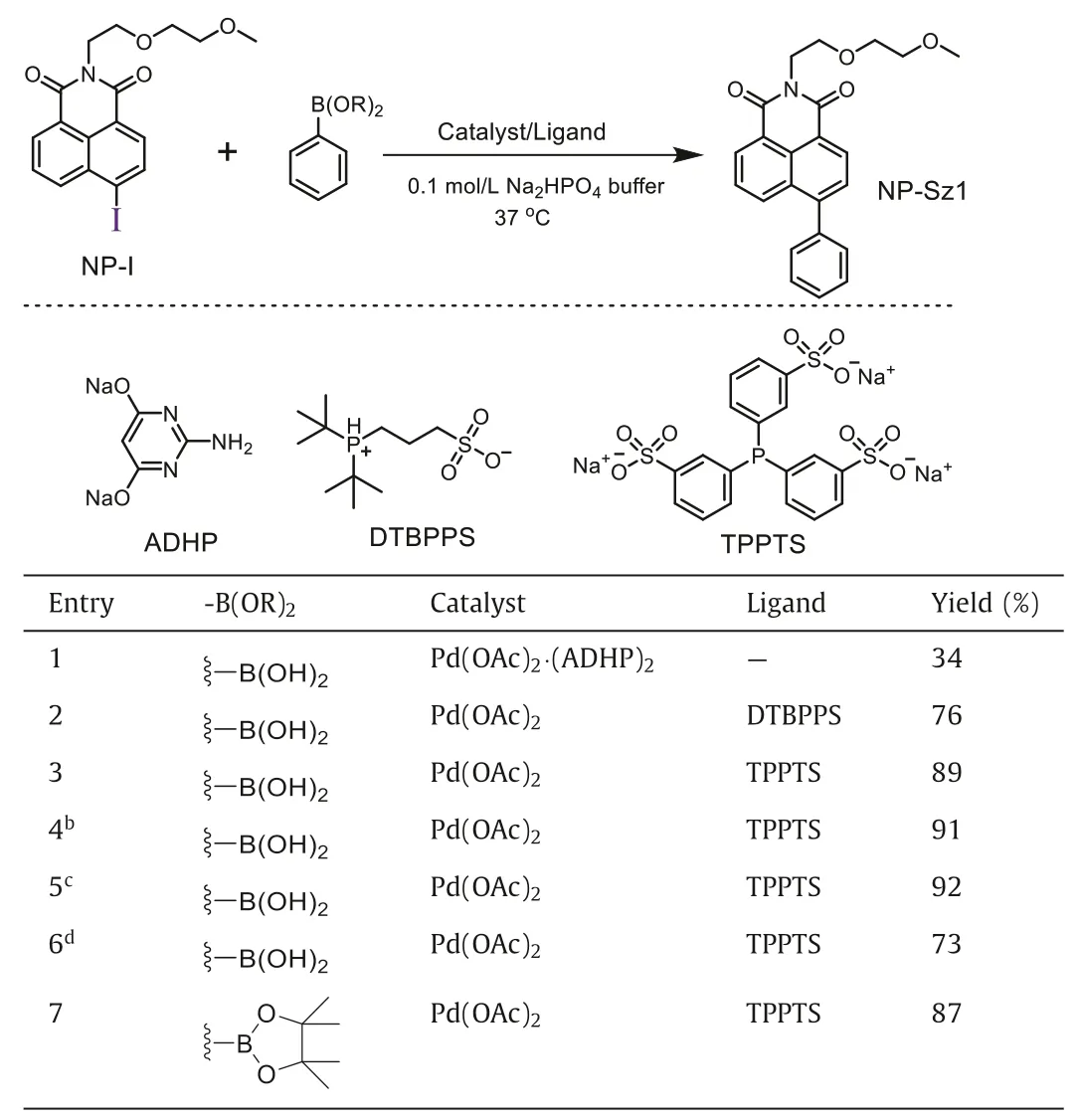
Table 1 Optimization of bioorthogonal Suzuki-Miyaura reaction conditions.a
In the following study, we investigated the influence of various factors including temperature, pH, viscosity, and redox status on this cross-coupling reaction (Figs.S1-S4 in Supporting informaton).We found that the reaction kinetics was slowed down at lower temperature (25°C,k2=0.24 L mol−1s−1), or in basic condition (pH 10.0,k2=0.071 L mol−1s−1), or in high-viscosity solution (50% glycerol aqueous solution,k2=0.058 L mol−1s−1), or in the presence of oxidant (H2O2,k2=0.16 L mol−1s−1).Furthermore, the reaction could not take place in acidic condition (pH 3.0)or in the presence of reductant (TCEP), which may be ascribed to their interferences in the catalytic cycle and impacts on the palladium catalyst activity [28].
With the optimal reaction conditions in hand, we examined the scope of this Suzuki-Miyaura reaction to assemble fluorescent 4-aryl-1,8-naphthalimides.Various phenyl boronic acids/boronates bearing electron-donating or electron-withdrawing group (1–8)and heteroaromatic boronic acids/boronates (9–14) were investigated.Gratifyingly, all fourteen Suzuki-Miyaura coupling products(NP-Sz1–14) were prepared with high yield (>80%) (Table 2), and distinctin situfluorescence enhancement in the reaction system was observed, which matches well with the emission of the purified coupling products (Figs.1a-c and Fig.S5 in Supporting informaton).Meanwhile, the reaction kinetics of NP-I with 9, 10, 11, 12,and 14 was found to be fast (k2ranging from 1.9 L mol−1s−1to 32.5 L mol−1s−1, Fig.S6 in Supporting information).
Next, photophysical assessment of these coupling products was performed.NP-Sz1–6 displayed absorption maxima around 350 nm and large Stokes shifts (Fig.S7 in Supporting information, Table 3).For non-substituted phenyl analog, NP-Sz1, 261-fold fluorescence enhancement at 415 nm emission wavelength (blue color)was observed (Fig.1a).The methoxyl group substitution on the phenyl ring (NP-Sz2) induced red shifts of 80 nm (λem=495 nm)to green visible region and 5277-fold increase in fluorescence(Fig.1b), indicating that electron-donating group could alter CT energy and in turn shift the emission wavelength.Conversely,adding electron-withdrawing cyan group on the phenyl ring (NPSz3) caused an 18 nm blue shift in emission (λem=397 nm, Figs.1c and d), which clearly depends on the weakening of donor of the ICT system.Hydroxyl substitution further red-shifts the emission peak to 505∼511 nm in the green region, while the brightness depends on the substitution position (Fig.1e).Para–hydroxy substitution (NP-Sz6) showed higher fluorescence intensity and quantum yields (Фf=0.3) than those ofortho–hydroxy (NP-Sz4) andmeta–hydroxy (NP-Sz5) substitution, perhaps due to its linear D-π-A system.
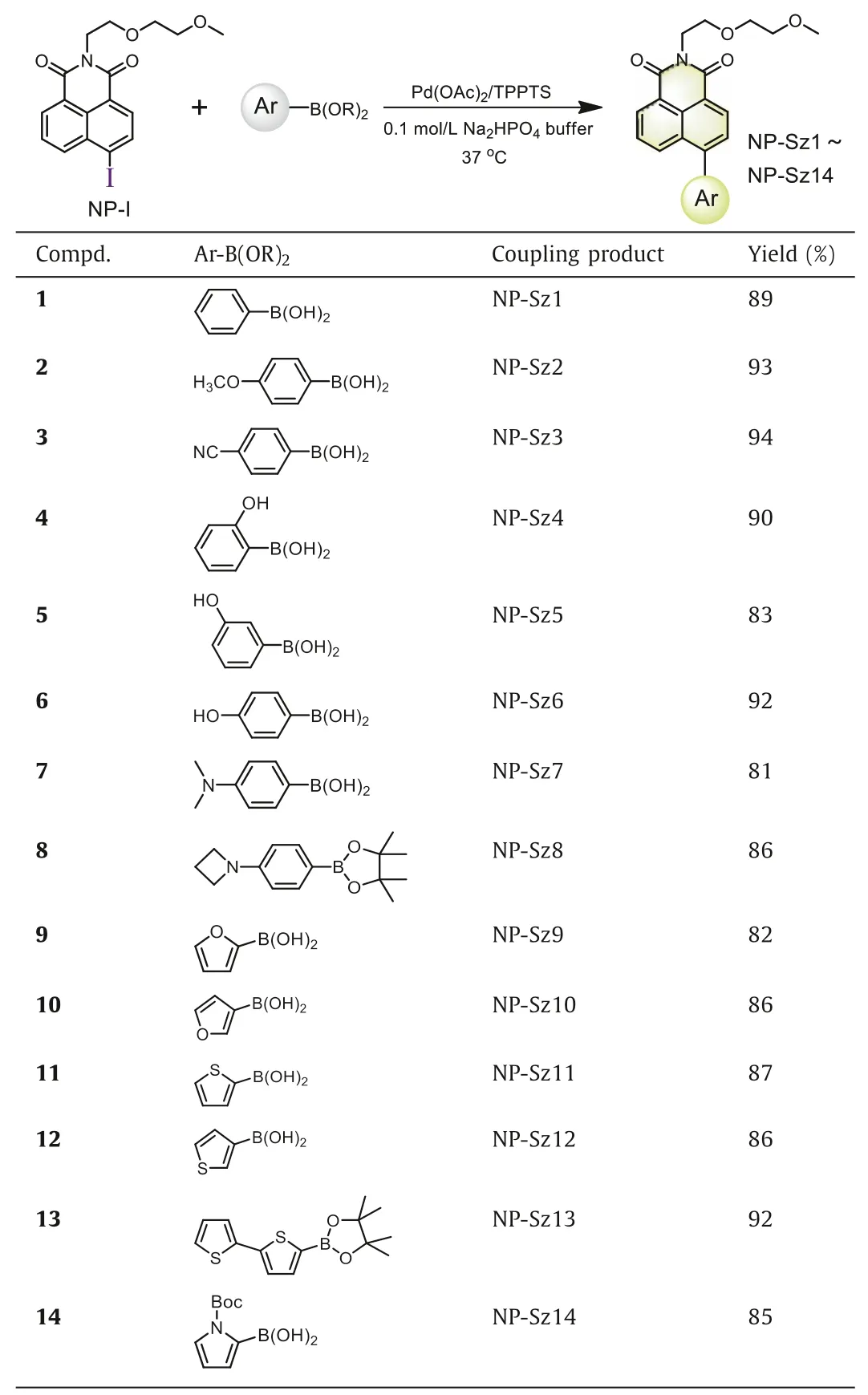
Table 2 Synthesis of 4-aryl-1,8-naphthalimides with Suzuki-Miyaura reaction.
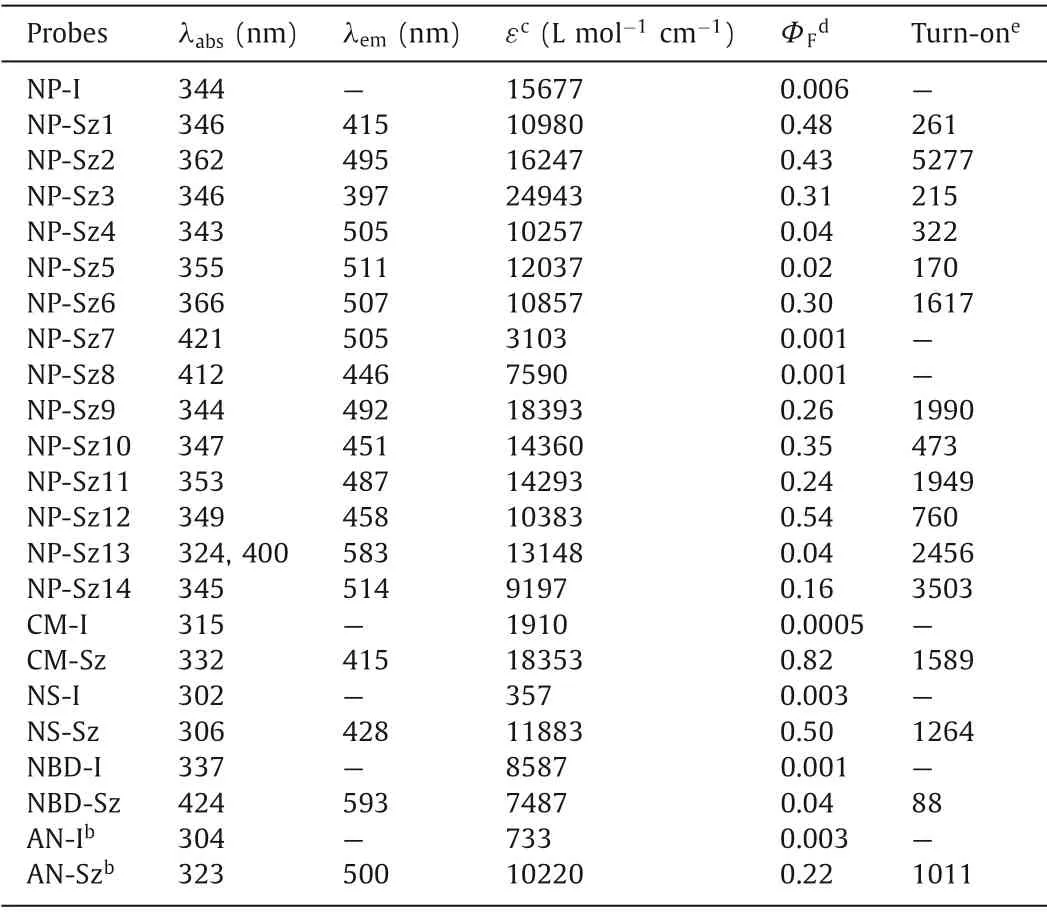
Table 3 Photophysical properties of ICT-based fluorogenic probes.a
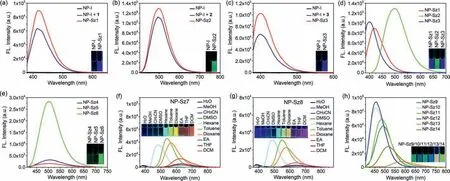
Fig.1.Fluorescence emission spectra and fluorescent images of NP-I and the in situ fluorescence enhancement after reaction with 1 (a), 2 (b), and 3 (c) in CH3CN.(d, e)Fluorescence emission spectra and fluorescent images of NP-Sz1–6 in CH3CN.Fluorescence emission spectra and fluorescent images of NP-Sz7 (f) and NP-Sz8 (g) in different solvents.(h) Fluorescence emission spectra and fluorescent images of NP-Sz9–14 in CH3CN.
Interestingly, replacement of methoxyl group with more electron-donating dimethyl amine group (NP-Sz7) or azetidine group (NP-Sz8) results in negligible fluorescence in acetonitrile.Since the solvent effects existed in most D-π-A molecules, we evaluated the emission spectrum of NP-Sz7 and NP-Sz8 in various solvents.As shown in Figs.1f and g and Tables S1 and S2(Supporting information), in strong polar solvents such as H2O,MeOH, CH3CN and DMSO, the fluorescence of NP-Sz7/8 was almost completely quenched.As the polarity of solvents increasing fromn-hexane to EA, the fluorescence intensity decreased and the emission color changed from cyan to red, revealing the twisted intramolecular charge transfer (TICT) effect of NP-Sz7/8 [29].
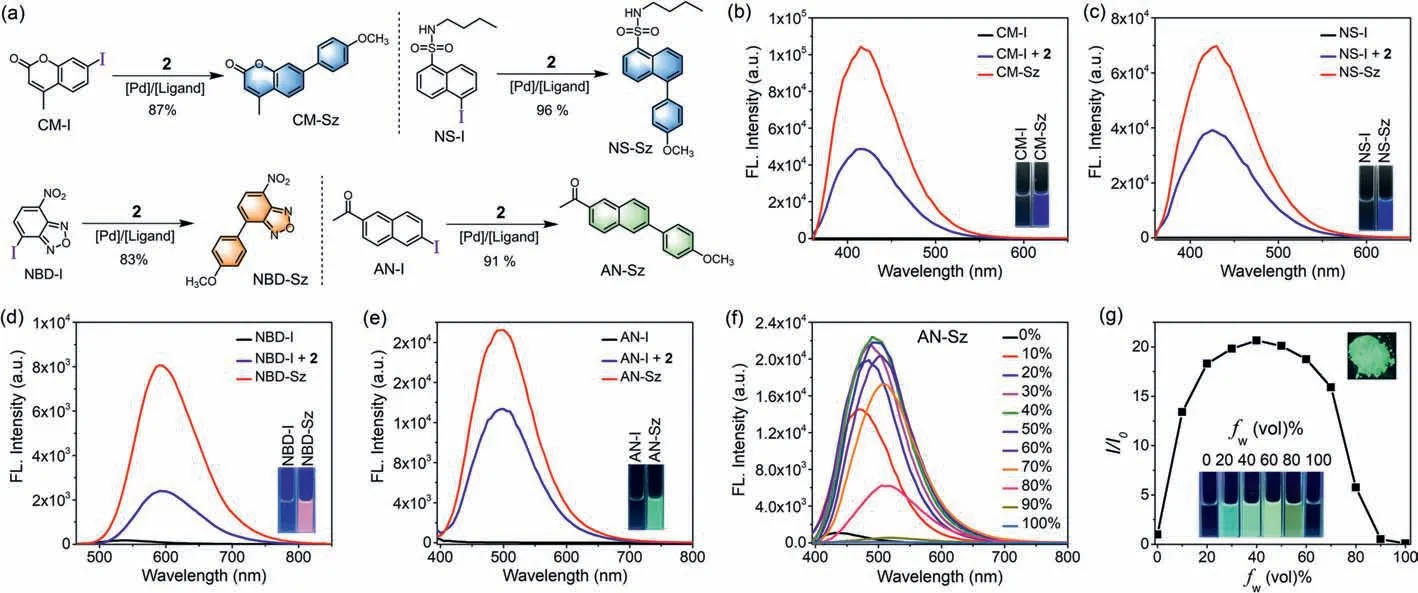
Fig.2.(a) Suzuki-Miyaura reaction between CM-I/NS-I/NBD-I/AN-I and boronic acid 2.(b-e) Fluorescence emission spectra and fluorescent images of 10 μmol/L CM/NS/NBD/AN probes in acetonitrile.(f) Fluorescence emission spectra of AN-Sz in CH3CN/H2O mixed solution (10 μmol/L) with different water fractions.(g) Plot of relative emission intensity (I/I0) of AN-Sz vs. water fractions, and its fluorescent image in solid state.
The five-member heteroaromatic ring substituted analogs also exhibited clear structure-property relationship (Fig.1h).The naphthalimide bearing 2-furyl (NP-Sz9) or 2-thienyl group (NP-Sz11) at 4-position displayed emission maxima around 490 nm in green region along with fluorescence quantum yield around 0.25.As comparison, 3-furyl (NP-Sz10) or 3-thienyl (NP-Sz12) substitution led to hypochromatic shift of emission and higher fluorescence intensity (Фf=0.35 and 0.54).In addition, introduction of Boc-protected 2-pyrrolyl group (NP-Sz14) gave rise to an emission peak of 514 nm and lower emission intensity (Фf=0.16).We also explored introducing a bithiophene group to further extend theπ-system (NPSz13).As expected, the emission wavelength was dramatically redshifted to 583 nm in the yellow-orange region.However, its fluorescence quantum yield is not satisfactory (Фf=0.04), demonstrating that fluorophores with long emission usually have lower brightness than that of short-wavelength fluorophores.
In the next study, we applied thisin situfluorogeneity strategy for designing other ICT-based light-up probes.Introduction of iodo group at the appropriate donor position afforded four emission quenched fluorophores, including 7-iodocoumarin (CM-I), 5-iodo-naphthalene-1-sulfonamide (NS-I), 4-iodo-nitrobenzoxadiazole (NBD-I), and 6-iodo-2-acetonaphthone(AN-I) (Fig.2a), for all of which significant fluorescence enhancement was observed upon Suzuki-Miyaura reaction with boronic acid 2, confirming the success of our design strategy.For coumarin and naphthalenesulfonamide fluorophore, the corresponding coupling products (CM-Sz and NS-Sz) achieved>1200-fold turn-on ratio around 420 nm emission wavelength in the blue region and extremely high fluorescence quantum yield (Фf=0.82 and 0.50,Figs.2b and c, Table 3).The nitrobenzoxadiazole coupling product(NBD-Sz) exhibited orange-red fluorescence (λem=593 nm) with relatively low fluorescence quantum yield (Фf=0.04) and turnon ratio (88-fold, Fig.2d).Notably, unlike other fluorophores, the coupling product of acetonaphthone (AN-Sz) was endowed with aggregation-induced emission (AIE) characters, which displayed bright fluorescence in the solid state (Figs.2f and g and Fig.S8 in Supporting information).Upon enhancing water fraction (fw)from 0 to 40% in the CH3CN/H2O mixed solution, gradual increase of fluorescence was observed, indicating that the AIE effect turns on the emission.A turn-on ratio of 1011-fold at emission maxima(λem=500 nm) and a fluorescence quantum yield of 0.22 were determined in the mixed solvents (fw=40%, Fig.2e).Further increasingfwfrom 40% to 100% results in obvious decreasing of fluorescence, presumably owing to TICT state being formed with an increase of polarity [30].
We then investigated the reaction between these aryl iodides and peptide.The boronic acid group was firstly introduced into the phenyl ring of phenylalanine from [D-Ala2, D-Leu5]-enkephalin, a kind of neuropeptide used for preventing neuronal damage against ischemic induced brain injury [31].The generated boronic acidcontaining enkephalin (BE) was then incubated with NP-I, CM-I,NS-I, NBD-I, and AN-I in the benign catalytic condition developed above.The LC-MS analysis identified the generation of coupling peptide after reaction for 8 h (Figs.S9-S14 in Supporting informaion), which validated the feasibility of this method in peptide modification.The slow reaction rate may be attributed to the formation of hydrogen-bond interaction between boronic acid and the C-terminal carboxyl acid in BE, which interferes the cross-coupling reaction.
To assess the feasibility of this fluorogenic approach for labeling proteins, NHS-activated NP-I (NP-I-NHS) was synthesized to modify bovine serum albumin (BSA) protein to produce NP-I conjugated BSA (BSA-NP-I), which was then incubated with various aryl boronic acids/boronates in Pd-mediated catalytic system and subsequently analysed by SDS-PAGE without further washing.As depicted in Fig.3a, robust signals were observed for the reaction of BSA-NP-I with all boron reagents, among which the reactions with 11 and 12 showcase strongest labeling efficiency, which are consistent with their high fluorescence quantum yields.Gratifyingly, unmodified BSA controls showed no detectable labeling, highlighting the specificity of this strategy.In order to achieve more specific target labeling, we applied this reaction for labeling of HaloTag protein, which could be conjugated with a functionalized haloalkane ligand based on the enzymatic ligation [32].A phenylboronic acid pinacol ester comprising a chloroalkane (15) was prepared and used for incubating with HaloTag protein at 37 °C for 2 h, affording boronate-conjugated HaloTag (HaloTag-B).Afterwards, HaloTag-B was reacted with different aryl iodides (NP-I, NBD-I, and AN-I)under catalytic condition for 12 h.Remarkably, the SDS-PAGE analysis revealed distinct fluorescent bands for the reaction of HaloTag-B with aryl iodides, while no emissive bands were noted in case of unmodified HaloTag (Fig.3b).Taken together, our results suggested that this Suzuki-Miyaura coupling strategy is an efficient tool in the application of fluorogenic protein labeling.
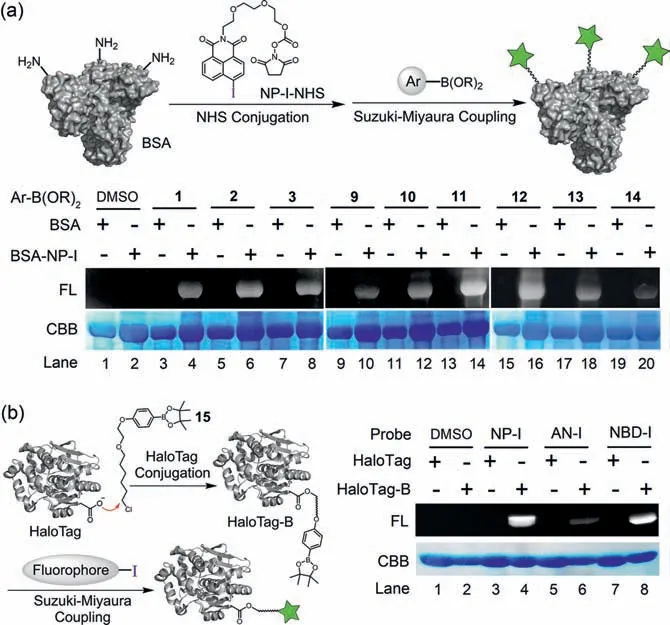
Fig.3.(a) Protein labeling of BSA-NP-I conjugate with boron reagents.(b) Protein labeling of HaloTag-B conjugate with aryl iodides.
Finally, we explored using this aryl iodide/boron reagent pair for fluorescent imaging of mitochondria in live cells.MTT study revealed that the boron reagents showed negligible toxicity against Hela cells (Fig.S15 in Supporting informaion).A mitochondrialocation probe, NP-I-TPP (Fig.4b), was synthesized and used for incubation with HeLa cells for 60 min.After removing the medium and washing briefly, the cells were treated with boron reagents 2/9/10/11/12/13 in presence of Pd(OAc)2/TPPTS, followed by confocal microscopy imaging without further washing.To validate the labeling selectivity of for mitochondria, cells were co-treated with MitoTracker Red (MTR), a commercially available mitochondria staining dye.As shown in Fig.4a and Fig.S16a (Supporting informaion), NP-I-TPP/boron reagents treatment produced crisp fluorescent mitochondrial images with an exceptional resolution, and excellent overlap of the signal between MTR and naphthalimide was observed (Figs.4c-f and Figs.S16b and c in Supporting informaion).Control experiments demonstrated that there is no specific staining of mitochondria nor background signal in the absence of boron reagents, therefore achieving a sharp contrast in fluorescent signal between the mitochondria and the background.Moreover,similar imaging results were found for HepG2 cells (Fig.S17 in Supporting informaion).Taken together, these results proved that this bioorthogonal Suzuki-Miyaura reaction was successfully applied for highly efficient and selective no-wash fluorescent staining of intracellular organelles in live cells in high target-to-background ratio.
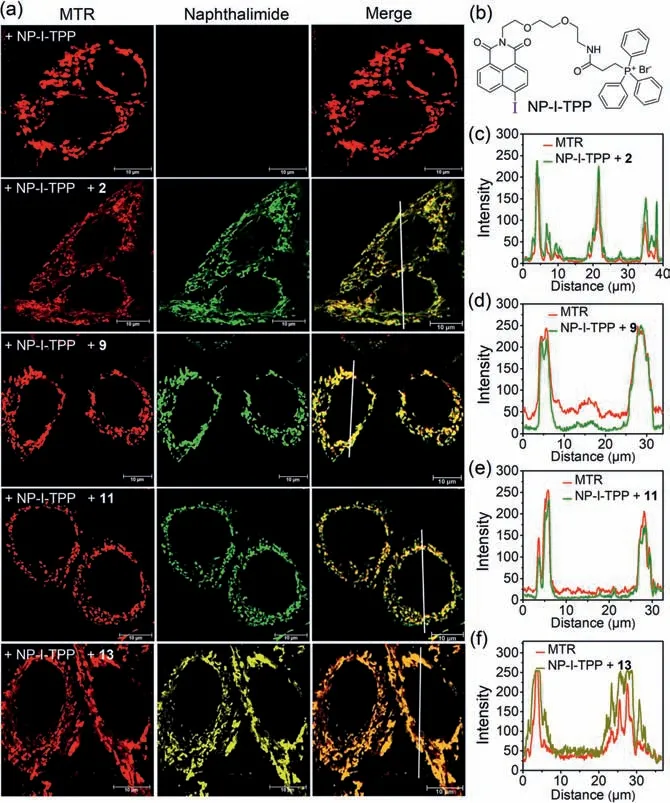
Fig.4.(a) Confocal images of Hela cells labeled by Suzuki-Miyaura reaction between NP-I-TPP and 2/9/11/13.(b) Structure of NP-I-TPP.(c-f) Fluorescence line intensity to measure the signal overlap of MTR (red) with naphthalimide (green or yellow).Scale bar=10 μm.
In summary, we described a general strategy forin situassembly of ICT-based light-up fluorophoresviabioorthogonal Suzuki-Miyaura cross-coupling reaction, which was applied for five fluorophore scaffolds including naphthalimide, coumarin, naphthalene sulfonate, nitrobenzoxadiazole, and acetonaphthone.The iodo group was used as the smallest bioorthogonal handle, which quenches the fluorescence by blocking ICT process when introduced at the appropriate position.Upon Suzuki-Miyaura reaction occurs, the multicolor fluorophores were assembledin situ, which exhibited notable fluorescence enhancement (up to 5277-fold turnon ratio) and high fluorescence quantum yields (up to 0.82).By thorough and systematic study of structure-property relationship of 1,8-naphthalimide based probes, we found that altering the substituents andπscaffold in the fluorophores allows fine-tuning of their photophysical properties.This iodo-boron bioorthogonal pair was successfully applied for peptides modification, fluorogenic protein labeling, and live-cell mitochondria imaging in high signalto-noise ratio without any further washing.This novel fluorogenic bioconjugation approach offers a pregnant expansion for the chemical biology toolbox, and provides a reliable design strategy for the development of high-performance bioorthogonal light-up probes for diverse biomedical application.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was supported by the Beijing Nova Program (No.Z201100006820049), the National Natural Science Foundation of China (No.21907109) and the CAMS Innovation Fund for Graduate Students (No.2019–1007–03).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2022.03.008.
 Chinese Chemical Letters2022年9期
Chinese Chemical Letters2022年9期
- Chinese Chemical Letters的其它文章
- A review on recent advances in hydrogen peroxide electrochemical sensors for applications in cell detection
- Rational design of nanocarriers for mitochondria-targeted drug delivery
- Emerging landscapes of nanosystems based on pre-metastatic microenvironment for cancer theranostics
- Radiotherapy assisted with biomaterials to trigger antitumor immunity
- Development of environment-insensitive and highly emissive BODIPYs via installation of N,N’-dialkylsubstituted amide at meso position
- Programmed polymersomes with spatio-temporal delivery of antigen and dual-adjuvants for efficient dendritic cells-based cancer immunotherapy