
基于单样本基因富集分析分型构建肺腺癌的风险预测模型
2022-09-14 11:46:24高晨吴林玉孔宁娄新璟郭勇许茂盛
浙江医学 2022年16期
高晨 吴林玉 孔宁 娄新璟 郭勇 许茂盛
肺癌是世界上发病率第二及死亡率最高的恶性肿瘤[1]。据研究报道,中国肺癌患者的5年总生存期仅为19.8%[2]。尽管手术方式、靶向治疗等技术的发展明显改善了肺癌患者的预后,但是仍然达不到预期效果[3-4]。肺腺癌是肺癌最常见的病理亚型,目前判断肺腺癌预后的主要依据是肿瘤的TNM分期,但这种方法存在较大的差异,精确性也有待提高[5-6]。多种分子机制参与了肺腺癌的发生、发展,只有对这些机制进行更深入的研究,才能更好地预测患者的预后,指导临床治疗,提高患者总生存期[7-8]。因此,临床上迫切需要寻找更为精准预测肺腺癌患者预后的分子生物标志物。本研究拟通过基于单样本基因富集分析(single sample gene set enrichment analysis,ssGSEA)的方法对来自癌症基因组图谱(The Cancer Genome Atlas,TCGA)数据库及基因表达数据库(Gene Expression Omnibus,GEO)中肺腺癌患者转录组的数据进行系统性生物信息学分析并构建肺腺癌的风险预测模型,为临床医师判断肺腺癌患者总生存期提供辅助工具,同时为寻找潜在的靶向治疗药物提供参考依据。
1 资料和方法
1.1 数据获取及整理 从基因组数据共享(Genomic Data Commons,GDC)官方网站(https://portal.gdc.cancer.gov)以及GEO官方网站(https://www.ncbi.nlm.nih.gov/geo)获取TCGA数据库及GSE26939数据集肺腺癌患者的转录组数据以及相关患者临床数据。TCGA肺腺癌数据库有515例患者的594个样本,其中59个为正常样本/癌旁样本,535个为肿瘤样本。GEO数据集作为外部验证集,数据集中有116例肺腺癌患者的116个肿瘤样本。从免疫学数据库和分析门户(The Immunology Database and Analysis Portal,ImmPort)网站(https://www.immport.org/home)共下载免疫相关的基因1 793个。从顺反组数据浏览器(Cistrome Data Browser,Cistrome DB)转录因子网站(http://www.cistrome.org)共下载转录因子318个。
1.2 ssGSEA及免疫分析 对来自TCGA数据库肺腺癌患者的转录组数据,运用ssGSEA分析及聚类分析,从而获得样本的免疫评分及不同免疫评分组;运用ESTIMATE分析,获得肿瘤样本的肿瘤微环境分数,并进一步分析肿瘤微环境分数在不同免疫评分组中的差异。运用CIBERSORT算法计算样本中22种肿瘤浸润免疫细胞的比例,并分析不同免疫评分组中肿瘤浸润免疫细胞比例的差异。在GSEA软件(版本4.1.0)上基于不同免疫评分组进行GSEA富集分析,获得显著的富集通路,并分析患者肿瘤样本的基因表达量在不同免疫评分组中的差异,获得在不同免疫分型组中的差异表达基因。通过取差异表达基因列表与免疫相关基因列表的交集,获得存在差异的免疫基因。
1.3 预后基因的筛选 在得到TCGA与GEO数据集中差异免疫基因表达量后,对其进行批次矫正。同时对患者进行进一步筛选,纳入标准:(1)病理检查确定为肺腺癌;(2)具有可获取的转录组数据;(3)具有可获取的临床数据。排除标准:(1)未同时具有可获取的转录组数据及临床数据;(2)缺少相关生存数据。最终确定纳入下一步研究的肺腺癌患者,其中TCGA数据库504例,GEO数据库115例。将来自TCGA数据集中的患者以7∶3随机分为训练集356例和内部验证集148例。同时将GEO数据集的患者作为外部验证集。基于训练集的数据,通过单因素Cox回归分析,筛选出肺腺癌患者预后相关的差异免疫基因。将预后相关的差异免疫基因与转录因子进行相关性分析,以及在Cytoscape软件(版本3.8.2)上进行相关蛋白互作网络的分析。
1.4 预后模型的建立 通过套索算法(least absolute shrinkage and selection operator,Lasso)回归对筛选出的肺腺癌患者预后相关差异免疫基因进行降维,并利用多元逐步Cox回归模型筛选出最优的差异免疫基因集合构建肺腺癌的风险预测模型,并获得每个样本的风险分数(Riskscore)。基于获得Riskscore的中位数,将患者分为高风险组及低风险组。运用内部及外部验证集的数据对预测模型进行检验,并采用ROC曲线及校正曲线来显示预测模型在训练组和验证组中预测模型的效能。采用Kaplan-Meier法对训练集、内部验证集、外部验证集进行生存分析。
1.5 联合模型及列线图构建 运用单因素及多因素Cox风险回归,将上述获得的Riskscore与患者的临床特征(性别、年龄、肿瘤分期)进行独立预后分析,获得肺腺癌患者独立预后因子并构建联合模型。采用ROC曲线、校正曲线及列线图分析该联合模型的效能及临床实用性。
1.6 统计学处理 采用R 4.0.5统计软件。运用的R语言包有 GSVA、limma、GSEABase、sparcl、Rtsne、ggplot2、estimate、pheatmap、reshape、reshape2、ggpubr、preprocessCore、venn、sva、survival、ggalluvial、dplyr、caret、glmnet、survminer、timeROC、rms、ggExtra、tidyverse、regplot。差异分析使用Wilcoxon秩和检验,相关性分析采用Pearson相关。P<0.05为差异有统计学意义。
2 结果
2.1 免疫评分及相关差异分析 根据肿瘤样本的ssGSEA免疫评分及聚类分析结果,将535个肿瘤样本分为高免疫评分组313个和低免疫评分组222个。高免疫评分组、低免疫评分组与肿瘤微环境分数的相关性见图1a(插页),其中高免疫评分组中的肿瘤纯度较低免疫评分组低,基质分数、免疫分数及ESTIMATE分数较低免疫评分组高,见图1b(插页)。运用Wilcoxon秩和检验分析基于CIBERSORT算法的12种肿瘤浸润免疫细胞的比例在高免疫评分组和低免疫评分组中的差异有统计学意义,见图1c(插页)。在高免疫评分组中显著富集通路有“cytokine-cytokine receptor interaction”“natural killer cell mediated cytotoxicity”“cell adhesion molecules”“T cell receptor signaling pathway”和“chemokine signaling pathway”等,见图2a(插页)。根据差异分析,在高免疫评分组和低免疫评分组中的差异表达基因有1 447个,见图2b(插页)。将差异表达基因与从ImmPort网站获取的免疫相关基因取交集后,获得免疫相关的差异表达基因有382个,见图2c(插页)。

图1 肺腺癌患者免疫分析及不同免疫评分组的差异分析(a:肿瘤纯度、ESTIMATE分数、免疫分数及基质分数与高免疫评分组和低免疫评分组之间的相关性,高免疫评分组中肿瘤纯度较低免疫评分组低,基质分数、免疫分数及ESTIMATE分数较低免疫评分组高;b:基质分数、免疫分数及ESTIMATE分数在高免疫评分组和低免疫评分组间差异有统计学意义,高免疫评分组的3个分数均比低免疫评分组高;c:基于CIBERSORT算法的12种肿瘤浸润免疫细胞的比例在高免疫评分组和低免疫评分组中差异有统计学意义)
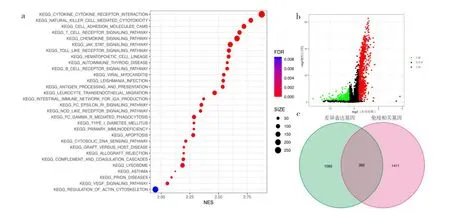
图2 GSEA富集分析及差异免疫基因确定(a:高免疫评分组中前5个显著富集通路有“cytokine-cytokine receptor interaction”“natural killer cell mediated cytotoxicity”“cell adhesion molecules”“ T cell receptor signaling pathway”和“chemokine signaling pathway”;b:火山图显示了在高免疫评分组和低免疫评分组中差异表达基因,绿色的是在高免疫评分组中低表达的基因,红色的是在高免疫评分组中高表达的基因;c:韦恩图显示了绿色的是1 447个差异表达基因,粉红色的免疫基因是1 793个,交集后免疫相关的差异表达基因有382个)
2.2 确定预后相关的差异免疫基因及其相关性分析 在对TCGA数据集及GEO数据集的转录组数据进行批次矫正且将其与上述免疫相关的差异表达基因列表取交集后,共得到219个差异表达基因的转录组数据。经过进一步筛选,共纳入619例肺腺癌患者,男287例,女332例,年龄33~90(65.04±10.20)岁,患者基线资料见表1。在训练集中,采用单因素Cox风险回归模型对219个基因的转录组数据及生存信息进行分析后,得到31个预后相关的差异免疫基因,见图3a(插页),这些预后相关的差异免疫基因与转录因子的相关性以及蛋白互作网络关系见图3b、c(插页)。

表1 TCGA及GEO数据集中肺腺癌患者临床资料
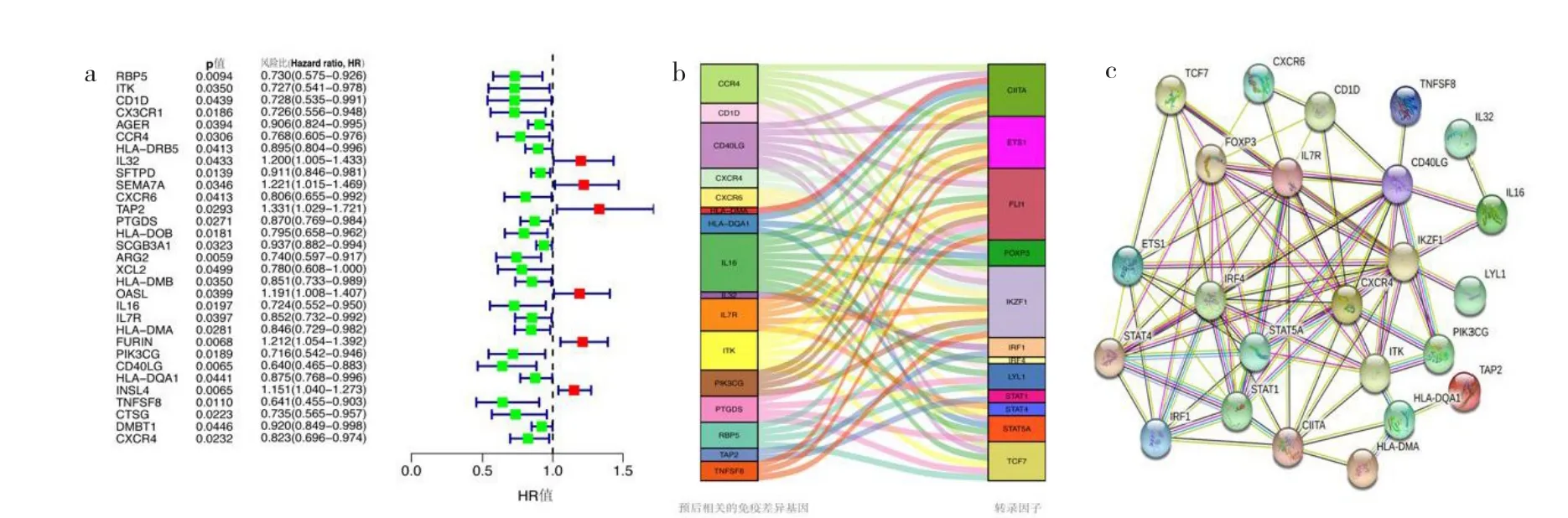
图3 预后相关的差异免疫基因及与转录因子相关性分析[a:森林图显示基于单因素Cox风险回归分析获得的31个预后相关的差异免疫基因;b和c:桑基图(b)显示与转录因子具有显著相关性的16个预后相关差异免疫基因(相关系数≥0.5,P<0.01),PPI图(c)显示了其蛋白互作的关系]
2.3 预后模型的构建及生存分析 经过Lasso回归及多元逐步Cox风险回归分析降维后,得到最优的差异免疫基因数据集并建立风险预测模型以及得到Riskscore,并根据训练集数据Riskscore的中位数,将训练集、内部验证集及外部验证集的患者分为高风险组及低风险组,其中训练集高风险组178例,低风险组178例;内部验证集高风险组76例,低风险组72例;外部验证集高风险组50例,低风险组65例。该模型的公式如下:Riskscore=EXP[(-0.211 434 889)×CX3CR1+(0.293 765 44)×IL-32+(-0.071 165 091)×SFTPD+(-0.333 423 936)×CXCR6+0.419 839 844×TAP2+(-0.269 368 749)×HLA-DOB+(-0.374 908 714)×ARG2+0.178 859 695×FURIN+(-1.040 515 441 267 49)]。该风险预测模型在对训练集及两个验证集患者的生存预测上均具有较好的表现,预测患者5年总生存期的AUC分别为0.703、0.713、0.750,见图4a-c(插页)。同时校正曲线分析显示该模型预测的1、3、5年患者总生存期都与实际总生存期较为一致,见图4d-f(插页)。此外,高风险组和低分险组患者的总生存期在3个数据集中差异均有统计学意义,见图4g-i(插页)。
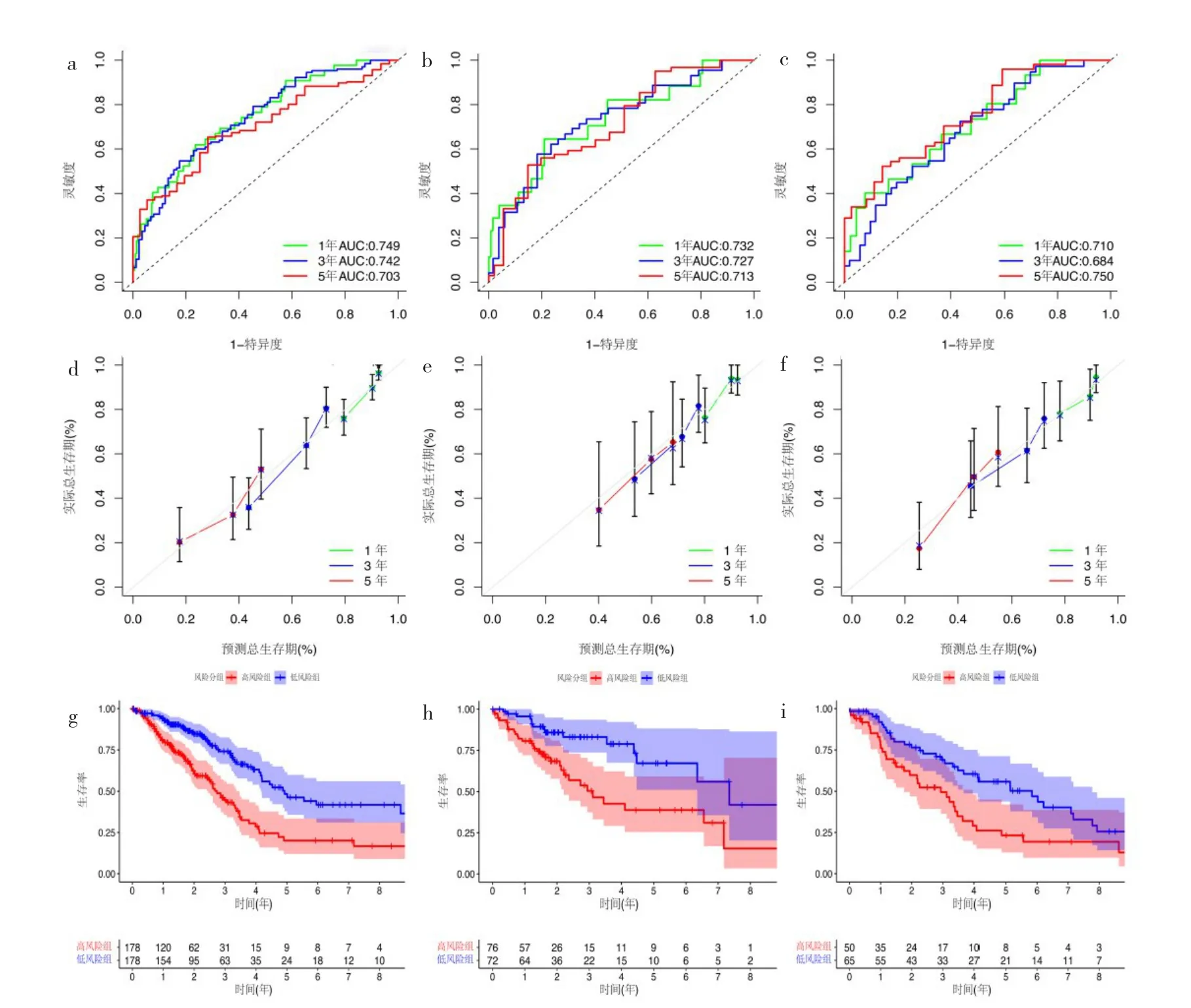
图4 预测模型的ROC曲线、校正曲线分析及生存分析[a-c:训练集、内部验证集、外部验证集的预测模型ROC曲线;d-f:训练集、内部验证集、外部验证集的校正曲线分析,结果显示预测结果与实际结果较为一致;g-i:在训练集、内部验证集、外部验证集中,高风险组和低风险组患者生存曲线差异均有统计学意义(均P<0.01),低风险组的患者总生存期较高风险组高]
2.4 独立预后分析及列线图的构建 经单因素及多因素Cox风险回归的分析结果显示,肺腺癌的分期及预测模型的Riskscore可作为肺腺癌患者的两个独立预后因子,见表2。肺腺癌的分期联合预测模型的Riskscore构建联合模型的1、3、5年总生存期的AUC分别为0.789、0.763、0.746。校正曲线分析显示该模型预测的1、3、5年患者总生存期与实际总生存期较为一致。同时该联合模型临床应用的列线图见图5。
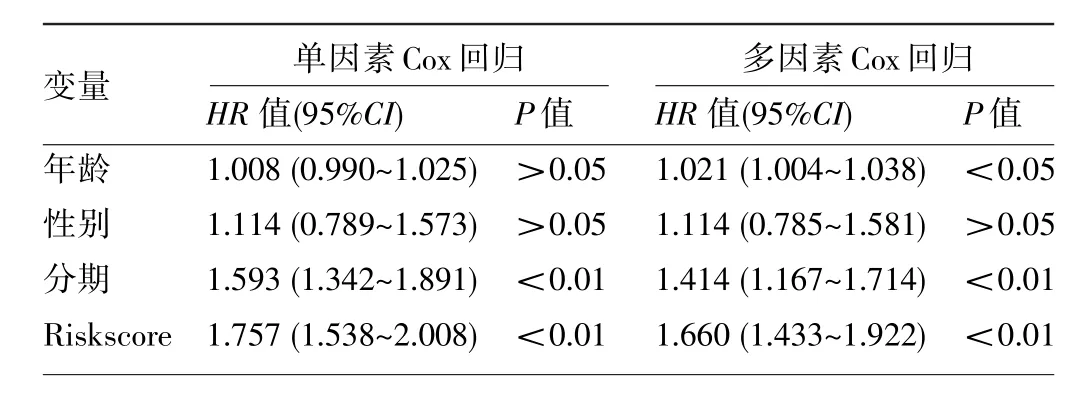
表2 单因素及多因素Cox回归分析
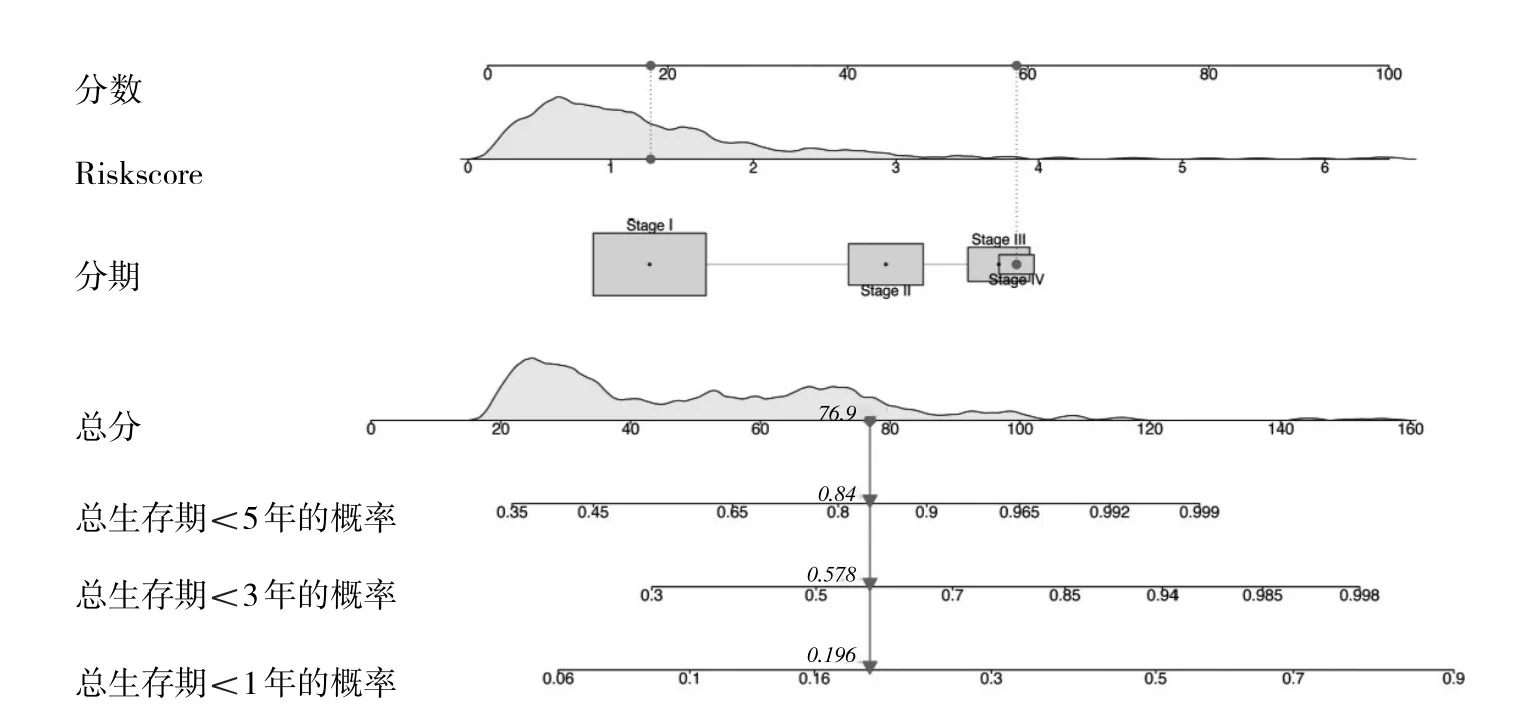
图5 联合模型的列线图的构建(点表示1例患者,其Riskscore与分期Ⅳ期的总得分为76.9分,总生存期<1年的概率是0.196,<3年的概率是0.578,<5年的概率0.84)
3 讨论
ssGSEA是能对单个样本的通路富集情况进行量化并评分的分析方法,例如免疫相关通路等[9]。同时已有不少研究报道了肿瘤微环境中免疫细胞浸润水平与肿瘤的发生、发展以及患者预后均相关[10-11]。而基于ssGSEA免疫分型来分析免疫相关差异表达基因在肺腺癌预后方面价值的研究尚缺乏。
本研究基于TCGA数据集中肺腺癌患者样本基因表达数据的ssGSEA免疫评分及Cox风险回归分析,获得肺腺癌预后相关的免疫差异表达基因。然后进行Lasso回归及多元逐步Cox风险回归分析降维,构建肺腺癌预后预测模型获得Riskscore,并用GEO数据集进行验证。利用Riskscore预测患者5年总生存期的AUC值均>0.7。将Riskscore与临床特征进行独立预后因子分析,并建立列线图,列线图1、3、5年的AUC也均>0.7。上述结果表明Riskscore及列线图在预测肺腺癌总生存期上都具有良好的效能。
本研究结果显示肺腺癌高免疫评分组的富集通路主要在“cytokine-cytokine receptor interaction”“natural killer cell mediated cytotoxicity”和“cell adhesion molecules”。而国内外也已有文献报道这些通路在非小细胞肺癌中的发生、发展中起重要作用[12-14]。例如,Zheng等[12]研究结果表明在高免疫浸润组的肺腺癌肿瘤样本也主要富集在“cytokine-cytokine receptor interaction”。Zhang等[13]研究也发现EPHA5突变会破坏自然杀伤细胞介导的细胞毒性对非小细胞肺癌的作用,促进癌细胞迁移。Li等[14]研究表明高免疫评分组和低免疫评分组的差异基因富集通路也包括“cytokine-cytokine receptor interaction”和“cell adhesion molecules”。上述研究结果证实了这些通路可以作为肺腺癌潜在的免疫治疗新靶点,进而提高肺腺癌患者预后水平。
本研究中,在对预后相关的免疫差异表达基因进行Lasso回归及多元逐步Cox风险回归分析降维后,获得了最优的8个基因集合,其中权重最高的3个基因分别是TAP2、ARG2和CXCR6。这些基因在非小细胞肺癌以及肺腺癌进展方面的作用也已经有一些研究报道[15-17]。Liu等[15]研究结果表明TAP2的基因多态性与非小细胞肺癌的进展存在潜在的关系。Giatromanolaki等[16]研究结果发现ARG2主要在癌相关成纤维细胞中表达。说明这些基因具有预测肺腺癌患者预后的能力。但是,也有研究结果发现癌细胞中表达的CXCR16并没有表现出对非小细胞肺癌预后的影响[17]。这可能是该研究纳入的是非小细胞肺癌患者,与本研究中纳入的肺腺癌患者不同所致,这需要进一步深入的研究来验证。
通过类似的方法,Wang等[18]研究也是基于ssGSEA算法分析出的免疫相关差异表达的长链非编码RNA(lncRNA)来构建肺腺癌预后的预测模型,其训练集预测5年总生存期的AUC达到0.63,验证集预测5年总生存期的AUC达到0.667。本研究则基于ssGSEA分析构建肺腺癌患者风险预测模型并获得Riskscore,其在训练集、内部验证集及外部验证集5年总生存期的AUC分别为0.703、0.713、0.750。同时将该Riskscore与临床特征进行独立预后分析并构建联合模型列线图,列线图5年总生存期的AUC达到0.746。Riskscore与列线图的AUC均较高且均>0.7,Riskscore的HR值也较临床分期高,均进一步表明了Riskscore在预测肺腺癌预后方面的能力较好,同时该模型也具有较好的泛化性。通过校正曲线分析也进一步显示了预测模型的结果与实际总生存期较为一致。
本研究存在一定的局限性。首先,研究纳入的患者例数有限,仅用了来自于TCGA数据库及GEO数据集,未来还需要多中心的数据来进行验证。其次,本研究并未对筛选的风险基因进行相关机制研究,未来需开展基础实验进行进一步分析。
综上所述,本研究基于ssGSEA分析获得的免疫相关差异表达基因构建了肺腺癌风险预测模型,并联合临床特征绘制了列线图,以期能够辅助临床医师对肺腺癌患者总生存期进行判断。
猜你喜欢
黄河之声(2022年10期)2022-09-27 13:59:46
中学生数理化(高中版.高二数学)(2022年4期)2022-05-25 13:08:00
中学生数理化(高中版.高二数学)(2022年4期)2022-05-25 13:08:00
中成药(2018年7期)2018-08-04 06:04:18
中学生数理化·八年级物理人教版(2017年11期)2017-04-18 11:22:51
西南国防医药(2016年6期)2016-12-01 06:01:08
癌症进展(2016年11期)2016-03-20 13:16:04
西南军医(2016年3期)2016-01-23 02:17:47
哈尔滨医药(2015年6期)2015-12-01 03:58:17
中国当代医药(2015年17期)2015-03-01 02:03:36
