
锌-钴氰化络合物催化剂的改性及其在聚醚碳酸酯二元醇合成中的应用
2022-09-09 02:23:12吴冬凡张源萍李其峰亢茂青王军威
燃料化学学报 2022年8期
吴冬凡 ,张源萍 ,李其峰 ,亢茂青 ,王军威
(1.中国科学院山西煤炭化学研究所 山西 太原 030001;2.中国科学院大学 北京 100049)
伴随着工业和社会的发展,全球二氧化碳的排放量日益增加。作为最主要的温室气体,大气中二氧化碳含量的增加加剧了“温室效应”,导致冰川融化、气候变暖、土地干涸等自然问题。直到2020年“双碳”目标的提出,人们更加深刻认识到二氧化碳固定与转化的重要性[1]。转化与利用的方式之一就是二氧化碳与环氧化物的共聚反应,将二氧化碳固定为高分子材料[2-4]。根据是否使用链转移剂,二氧化碳与环氧丙烷反应可以合成高分子量的聚碳酸酯和低分子量的聚醚碳酸酯二元醇[5-8]。其中,低分子量聚醚碳酸酯二元醇可以进一步和异氰酸酯反应合成聚氨酯,如式(1)所示。秦玉升等[9]以稀土掺杂的Zn3[Co(CN)6]催化剂在链转移剂存在时通过环氧丙烷和二氧化碳共聚,得到聚醚碳酸酯多元醇前体;在路易斯催化剂下,将聚醚碳酸酯多元醇前体和PO在60–150 ℃下封端,得到高伯羟基含量的聚醚碳酸酯多元醇,并利用该聚醚碳酸酯多元醇制备出性能优异的水性聚氨酯[10,11]。安娜[12]成功合成出一系列不同相对分子质量、CO2含量及官能度的聚醚碳酸酯多元醇并将其应用于聚氨酯弹性体的制备中,所得聚氨酯弹性体具有较好的力学性能。胡曦华[13]采用改性后的DMC催化剂,成功的使小分子多元醇引发反应,制备出分子量相对可控的聚氧化丙烯多元醇,且反应诱导期较短,结果低于其他文献报道。此外江苏钟山化工有限公司、上海佳化化学有限公司以及万华化学有限公司等也在开发聚醚碳酸酯多元醇的工业化应用。因聚醚碳酸酯多元醇广泛的应用前景,而逐渐成为研究热点。
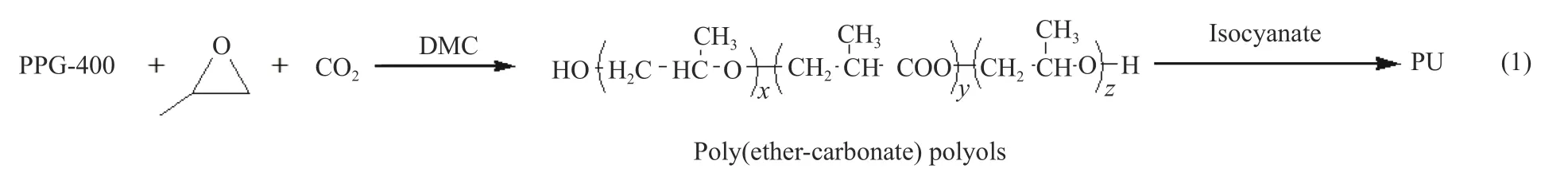
由于CO2具有较高的热力学稳定性不易插入到聚合物链上,为了促进聚合反应顺利进行,提高CO2转化率,催化体系选择尤为重要。目前,用于该反应的催化体系有二乙基锌–多质子催化体系、金属Salen催化体系、羧酸锌催化体系、稀土金属盐催化体系和双金属氰化物(DMC)催化体系等[14,15]。其中,DMC催化剂具有活性高、用量少、易制备等优点而得到广泛应用[16],目前,关于其明确的催化机理尚不统一,大部分文献认为存在均聚、共聚、回咬生成副产物三种过程,如图1所示。

图1 DMC催化剂反应机理[12]Figure 1 Reaction mechanism of DMC catalyst[12]
有研究表明,DMC催化剂活性与结晶度密切相关[16],而通过加入配体、共配体可以降低结晶度。近年来,许多关于DMC催化剂配体及共配体的研究相继报道。An等[17]使用叔丁醇与Zn2+配位,形成基础立方晶体结构,再加入高浓度PPG-3000配体降低结晶度,从而提升了催化剂活性。Tran等[18]使用α-、β-、γ-和δ-二羰基络合剂为配体制备了Zn-Co双金属催化剂,该催化剂可制备出碳酸盐含量高、分子量范围宽、多分散性窄的聚碳酸酯多元醇,且合成的聚碳酸酯多元醇的功能取决于使用络合剂的类型。Meng等[19]在制备催化剂过程中使用了非离子表面活性剂NP-40,利用NP-40胶化作用促进催化剂表面与正电荷的吸附和相互作用,明显降低催化剂粒径进而提高活性。研究证明,有机化合物能够破坏立方晶体结构,降低结晶度,而且在DMC催化剂的成核和生长过程中,共配体中–OH、–NH2与Zn2+、Co3+发生络合作用,能维持催化剂结构稳定[20]。因此,本研究提出了共配体存在条件下DMC催化剂形成过程,如图2所示。
目前,在对DMC催化剂改性的相关报道中,多数集中在配体上,而对于共配体的选择报道较少,本研究通过加入共配体以达到两个目的:一是,降低结晶度,提高催化剂活性;二是,减小催化剂粒径。以叔丁醇为配体,选用不同结构、活性基团的Span80、Tween80、D400和D2000为共配体[21](如表1所示),系统研究了在双向滴加过程中,共配体种类对催化剂结晶度、粒径及活性的影响。
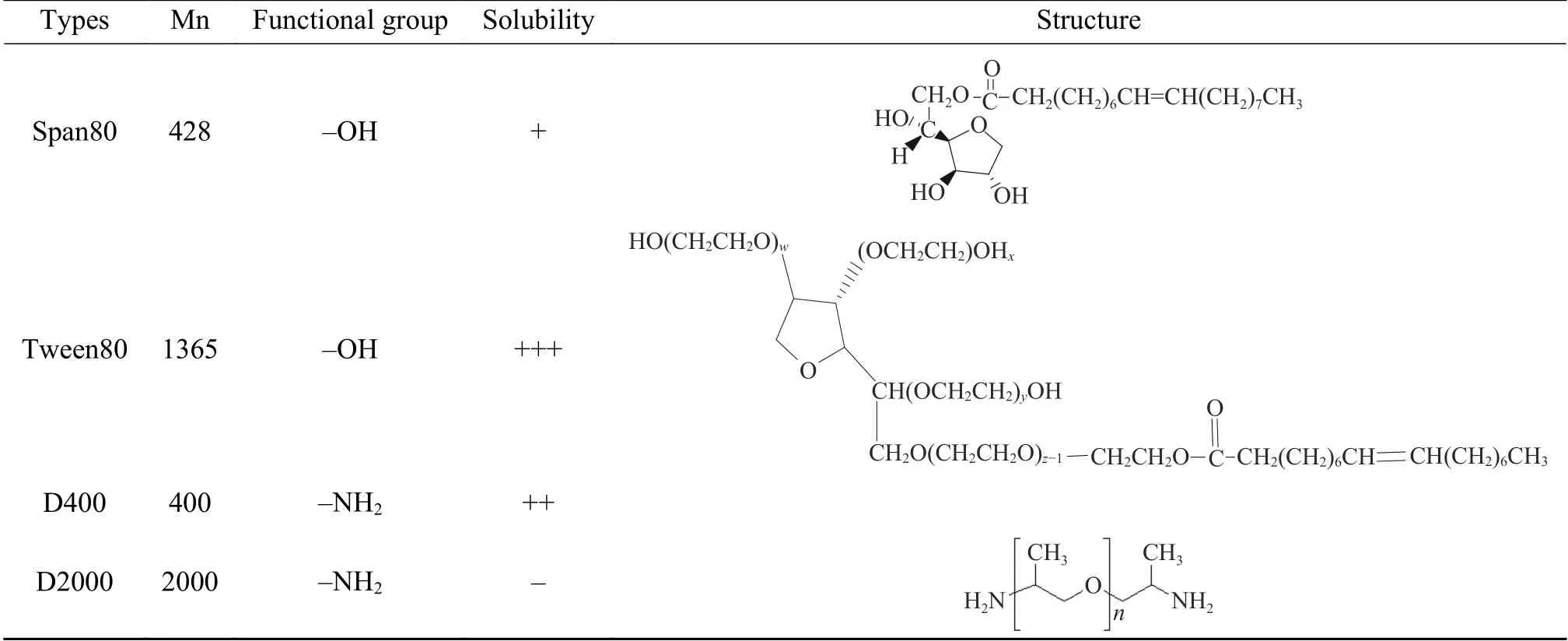
表1 共配体的种类及结构Table 1 Types and structures of co-complexing agents
1 实验部分
1.1 原料与试剂
氯化锌(ZnCl2,分析纯,国药集团化学试剂有限公司);六氰合钴酸钾(K3[Co(CN)6],分析纯,阿拉丁试剂(上海)有限公司);叔丁醇(t-BuOH,分析纯,天津科密欧化学试剂有限公司);失水山梨醇单油酸酯(Span80,工业级,青岛优索化学科技有限公司);失水山梨醇单油酸酯聚醚(Tween80,工业级,青岛优索化学科技有限公司);聚醚胺D400(阿拉丁试剂(上海)有限公司);聚醚胺D2000(阿拉丁试剂(上海)有限公司);环氧丙烷(PO,分析纯,国药集团化学试剂有限公司);二氧化碳(CO2,99.9%,山西宜虹气体工业有限公司)。
1.2 催化剂的制备
采用共沉淀法制备DMC催化剂。在快速搅拌条件下,将K3[Co(CN)6]的水溶液(8 g,140 mL)和ZnCl2溶液(25 g,40 mL H2O+20 mLt-BuOH)在1 h内均匀滴加到混合溶液(120 mL H2O+80 mLt-BuOH)中,滴加结束后室温继续搅拌1 h,然后转移至烧瓶,在机械搅拌条件下80 ℃保温3 h。最后化浆、洗涤、干燥、称重,得到DMC催化剂。改性的DMC催化剂制备方法是在原有制备方法基础上,滴加结束后分别加入Span80、Tween80、D400和D2000,搅拌1 h,后续保温、洗涤过程不变。最终得到四种添加不同共配体的催化剂,分别记作DMC+S80、DMC+T80、DMC+D400和DMC+D2000。
1.3 催化剂的表征
采用X射线粉末衍射技术(XRD,D8-ADVANCE德国Bruker公司)表征催化剂晶体结构以及结晶度变化,以CuKα光源进行辐射,2θ为5°–60°,扫描步长为0.02°;采用傅里叶变换红外光谱仪(Nicolet 380,美国Thermo electron)对样品进行结构测试,扫描为4000–400 cm-1;采用扫描电子显微镜(SEM,JSF-7001F 日本电子公司)观察样品表面形貌,加速电压为5 kV,测试前样品在丙酮中分散处理;采用激光粒度分析仪(Winner2005,济南微纳颗粒股份有限公司)考察催化剂在水溶液中的粒径及粒径分布。
1.4 催化剂活性评价
以PPG-400为链转移剂,环氧丙烷和二氧化碳共聚反应,根据聚合产物的理化性质评价催化剂活性。具体反应过程为:将链转移剂、催化剂加入到干燥的高压反应釜中,N2置换三次。然后升温至120 ℃,使用输液泵快速加入5 g PO待出现明显“温升压降”现象,降温至80 ℃,保持压力2.0 MPa,使用输液泵以10 g/h速率加入剩余PO,进料结束后继续反应2 h。待反应结束后,脱除残余单体,即得到聚合产物。利用氢核磁共振仪(1H NMR,AVANE 400 MHz,瑞士Bruker)计算聚合产物中二氧化碳含量,用乙酸酐法滴定产物羟值进而计算数均分子量[12]。采用凝胶渗透色谱(GPC,LC-20AD 日本岛津)测定聚合产物分子量分布。
2 结果与讨论
2.1 催化剂结构表征
图3是共配体改性后的催化剂FT-IR谱图,2192 cm-1、471 cm-1分别归属于DMC催化剂Co–CN–Zn结构中的–CN、Co–CN,3451 cm-1处宽峰是–OH的吸收峰,由于相邻–OH发生缔合导致吸收峰向低波数方向移动。以上特征峰在改性后的催化剂中均有出现,证明加入共配体不会破坏原有的配位结构。使用表面活性剂改性后的两种催化剂中,从红外谱图中可观察到特征衍射峰没有发生变化并且没有出现Tween80和Span80中羰基(–C=O)、脂肪环的吸收峰,表明,DMC+T80、DMC+S80与改性前DMC结构基本一致。可能是因为Tween80和Span80结构带有脂肪环,作为共配体位阻较大不能与催化剂骨架发生作用,所以没有改变催化剂晶体结构。而DMC+D400、DMC+D2000与改性前DMC结构相比,在3496 cm-1、3453 cm-1都出现明显的归属于伯胺(–NH2)的伸缩振动吸收峰,在1624 cm-1出现N–H面内弯曲振动吸收峰,表明催化剂中氨基的存在。通过对比这两类不同共配体改性后的催化剂红外谱图,作者发现加入的表面活性剂(Tween80和Span80)没有保留在催化剂体系中,可能在后续的洗涤过程中除掉了。而使用聚醚胺作为共配体的催化剂,结构上明显观察到D400、D2000官能团的存在,说明氨基与催化剂发生化学相互作用并保留在催化剂体系中。
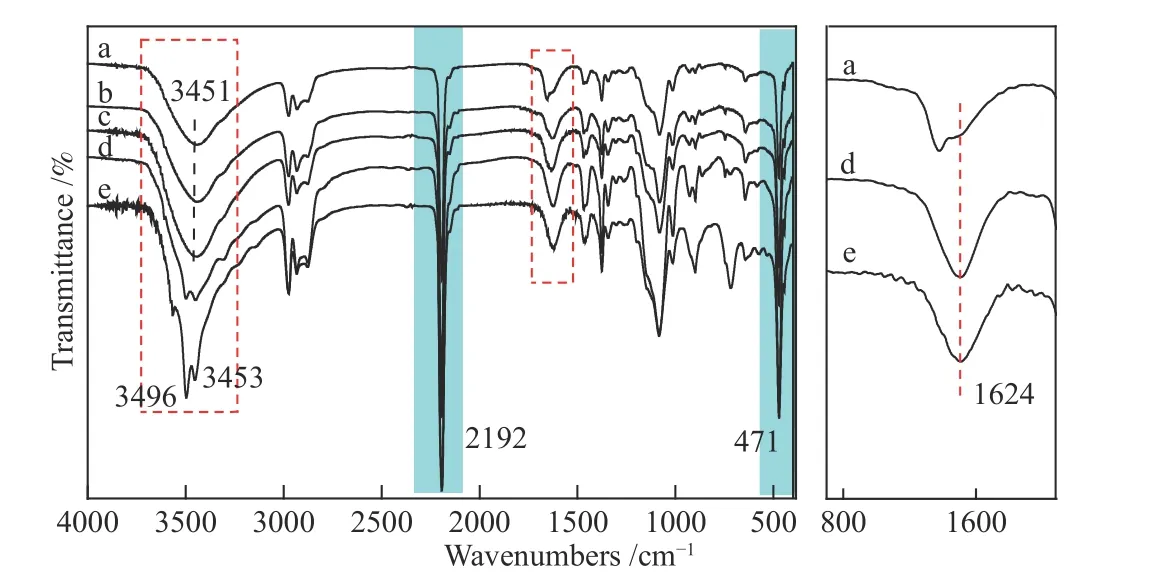
图3 不同共配体改性的DMC催化剂的FT-IR谱图Figure 3 FT-IR spectra of DMC catalysts modified by different co-complexing agents
将改性后的四种催化剂进行XRD表征,结果如图4。与未改性的DMC催化剂相比,DMC+S80和DMC+T80的衍射峰位置及强度并没有发生改变,表明Tween80和Span80加入后催化剂仍维持原有的晶体结构,这与红外谱图观察到的结果相符。聚醚胺的加入使催化剂晶体结构发生变化。在11.16°、30.28°–31.0°、32.77°–33.49°出现了三组新衍射峰,为Zn5(OH)8Cl2·H2O的特征峰[22,23],因为滴加阶段反应中ZnCl2是明显过量的,可能是未反应的ZnCl2小液滴水解后形成Zn5(OH)8Cl2·H2O被聚醚胺包裹起来,而留在了体系中。而且DMC+D400中Zn5(OH)8Cl2·H2O的 衍 射 峰 强 度 明 显 高 于DMC+D2000,可能是由于D400在水和叔丁醇的混合液中溶解性大于D2000,更容易影响催化剂结构。在17.4°(200)、24.7°(220)、35.2°(400)三个位置出现的衍射峰为Zn3[Co(CN)6]2·nH2O的特征吸收峰,表明DMC+D400具有明显的立方晶体结构[24]。
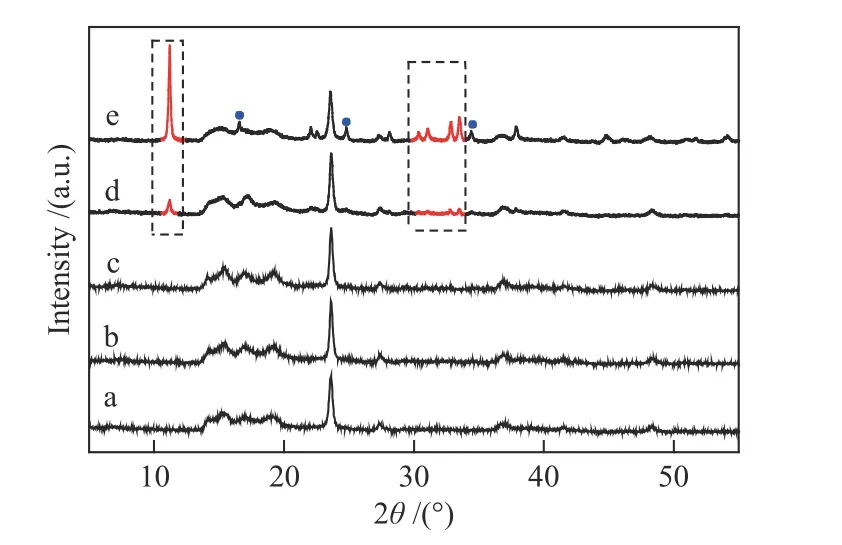
图4 不同共配体改性催化剂的XRD谱图Figure 4 XRD patterns of catalysts modified by different co-complexing agents
2.2 催化剂形貌表征
对改性后的催化剂进行粒径分析,如图5, DMC催化剂粒径分布不均且平均粒径较大为11.2 μ m,这是由于在双向滴加过程之前,先向ZnCl2水溶液中加入部分t-BuOH溶液,产生“预配位”作用[12],该过程可能会使ZnCl2小液滴粒径增加,从而影响整个催化剂粒径分布。加入不同配体改性后的催化剂平均粒径有明显变化。其中,经Tween80改性的催化剂粒径分布变宽,平均粒径大小(11.59 μ m)几乎没有变化;而Span80改性的催化剂分布变宽且平均粒径增大(21.41 μm),这是因为Span80具有极强的疏水性,经过较长时间保温过程使原有片层结构堆叠,进而形成更大的催化剂颗粒且分布不均。加入两种聚醚胺后,DMC+D400、DMC+D2000粒径分布变窄,且平均粒径减小(3.31 μm、5.11 μ m)。推测是在保温过程中,D400、D2000将催化剂小颗粒包裹起来,减少了催化剂颗粒之间的碰撞、聚集等相互作用,导致粒径分布较平均。
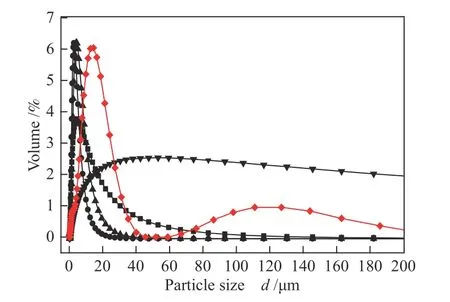
图5 不同共配体改性的催化剂粒径分布Figure 5 Mean volume diameter distribution of catalysts modified by different co-complexing agents
为了研究催化剂的表面形貌,对催化剂进行了扫描电镜的测试,如图6。催化剂形貌主要受双向滴加过程中滴加速率、滴加时间、搅拌速率等因素影响[12]。DMC催化剂形成的过程可分为以下阶段:当K3[Co(CN)6]和ZnCl2同时滴加到叔丁醇与水的混合溶液中,在反应初始阶段生成大小均匀的催化剂颗粒,此为成核过程;随着反应进行,受溶液体系限制,在高速搅拌下催化剂颗粒之间相互碰撞、吸附,此为生长过程;在保温过程中,叔丁醇配体以及共配体与催化剂骨架发生相互作用,破坏立方晶体结构增加无定形态结构;最后,经过多次的化浆、洗涤,去除未反应的K+、ZnCl2和多余配体。经该过程得到的DMC催化剂由同一尺寸小颗粒堆叠而成,有立体交错形成的“花朵”状结构和简单层叠形成的片状结构。“花朵”状结构能暴露出更多的活性位点,有利于催化剂活性提高。
在80 ℃保温过程中加入不同组成(Span80、Tween80、D400、D2000)的高浓度有机共配体,通过物理或化学相互作用增加“花朵”状结构,从而达到对催化剂改性的目的。从图6可知,使用Span80、Tween80改性后的催化剂,“花朵”状结构减少甚至消失,几乎都为片状结构。这是因为Span80、Tween80未保留在催化剂体系里,在催化剂形成过程中由于自身的物理作用改变了催化剂形貌。尤其是DMC+S80,因为Span-80具有极强的疏水性,经过较长时间保温过程使原本的立体“花朵”结构被“打散”,然后相互堆叠形成更多的片层结构。使用聚醚胺改性后的催化剂“花朵”状结构明显增多,可能是聚醚胺中活性基团 (–NH2)、氧原子与催化剂产生化学作用而改变催化剂形貌。与DMC+D400相比,DMC+D2000每一“簇”体积减小且“花瓣”之间距离增加,这有利于暴露出更多的活性位点[24],也易于环氧丙烷和CO2与催化剂接触,对提高催化剂活性有利。

图6 不同共配体改性的催化剂SEM照片Figure 6 SEM images of catalysts modified by different co-complexing agents
2.3 催化剂热稳定性测定
对五种催化剂进行热稳定性分析,得到热失重曲线,如图7。图7(a)、(c)为DMC、DMC+S80、DMC+T80的热失重曲线,三种催化剂失重过程相似,且最终残留量相近约为39.99%,进一步证明Span80和Tween80未保留在催化剂体系中。图7(b)、(d)为聚醚胺作为共配体改性的催化剂,与DMC相比,因聚醚胺保留在催化剂体系中,因此,DMC+D400 、DMC+D2000在150–450 ℃观察到更多的失重过程。DMC+D400催化剂结晶度较高,样品中配体及共配体含量相对减少,使最终样品残留量最高约为44.13%[22,25]。在评价催化剂活性的反应中,反应温度80 ℃,激活温度120 ℃,该温度下不会对催化剂结构产生影响,也证明激活温度设置在120 ℃对催化剂来说是合理的。

图7 不同共配体改性的催化剂TG和DTG曲线Figure 7 TG and DTG curves of DMC catalysts modified by different co-complexing agents
2.4 催化剂活性评价
评价催化剂活性,首先要确定合适的反应装置及反应条件, 不同条件下CO2与 PO共聚反应结果如表2所示。在CO2与 PO共聚反应中,可通过链转移剂调控聚醚碳酸酯二元醇的分子量[26,27]。链转移剂与环氧丙烷质量比越小,合成的聚醚碳酸酯二元醇分子量越大(entries 2、3、8)。低分子量聚醚碳酸酯二元醇反应大多采用一步法,即在t=80 ℃,p=2.0 MPa条件下,反应釜中一次性加入环氧丙烷、催化剂、链转移剂,反应20 h(entry 1)。该方法合成的聚醚碳酸酯二元醇分子量低、激活时间长,更重要的是体系中存在过量环氧丙烷,激活时环氧丙烷自聚形成聚醚并放出大量热,使温度、压力突增,这明显增加了对反应釜的要求,在工业应用过程中也存在极大的安全隐患,不利于将反应扩大至工业化。为了解决这一问题,在传统一步法基础上改进,使用连续进料法(entries 2–6)进行反应。连续进料法分为三个阶段,第一激活阶段:将链转移剂、催化剂加入反应釜中,升温至120 ℃,使用输液泵加入少量环氧丙烷,几分钟后观察到明显“温升压降”现象,证明DMC催化剂已被激活;第二进料阶段:降温至80 ℃,通入CO2至2.0 MPa,使用输液泵按一定速率加入剩余环氧丙烷;第三后反应阶段:进料结束后,保持t=80 ℃,p=2.0 MPa,继续反应2 h。与一步法相比,连续进料法催化剂激活时间缩短,反应时间缩短,CO2插入量增加,可以通过进料速率控制反应速率使整个过程稳定,该方法更适用于工业化。而且设计分子量为2000 g/mol的聚醚碳酸酯二元醇,采用连续进料法时产物分子量更接近于设计分子量(entries 1、2)。在CO2与 PO共聚过程中,温度的升高通常伴随着聚合物中碳酸酯含量的降低和副产物环状碳酸酯的增加(entries 5、6);温度的降低会导致催化剂激活后很难继续与PO反应,使产物分子量减小(entry 4)。结合已有文献及实验探究确定80 ℃为反应温度。

表2 不同条件下CO2与 PO共聚反应Table 2 Copolymerization of PO and CO2 catalyzed under different conditions
采用连续进料法评价四种催化剂活性,以合成分子量为2000 g/mol的聚醚碳酸酯二元醇为目标进行实验,结果如表3所示。催化剂活性越高,聚醚碳酸酯二元醇分子量越接近2000 g/mol。未改性的DMC催化剂合成产物的分子量为1204 g/mol,改性后的DMC+D2000催化剂合成产物的分子量增加至1679 g/mol,且碳酸酯单元质量分数增加,说明催化剂DMC+D2000活性提高。根据产物分子量大小,表明改性后的四种催化剂DMC+S80、DMC+D400、DMC+T80和DMC+D2000活性依次增加。原因分析如下:根据前文表征Span80、Tween80只是改变了催化剂的表面形貌,使改性后的催化剂多为片层结构,催化剂颗粒之间相互堆叠,导致PO、CO2不容易与催化剂活性中心接触,不利于共聚反应进行。与DMC+S80和DMC+T80相比,DMC+D2000的催化剂活性明显提高,这与聚醚胺结构中的氨基有关,一方面,–NH2与环氧丙烷的O原子通过氢键作用促使C-O键极化,有利于环氧化物开环[23],提高环氧丙烷转化率;另一方面,是因为D2000具有碱性,易于吸附CO2,提高CO2插入量。但是对DMC+D400来说,它的结晶度更高,催化剂活性变低。这是由于D400分子量小,体系中–NH2含量相对增加,吸附了更多的CO2,而CO2与环氧丙烷在催化剂的活性位点(Zn–O)相互竞争导致环氧丙烷转化率低。综上所述,加入D2000作为共配体改性后得到的催化剂——DMC+D2000,结晶度低,活性高。

表3 不同催化剂作用的CO2与 PO共聚反应aTable 3 Copolymerization of PO and CO2 catalyzed by different catalysts a
将DMC+D2000的催化性能与文献中的催化剂进行对比,结果见表4。DMC催化剂中引入的共配体从短链醇到高度支化聚合物再到长链聚醚胺,都对催化剂活性产生影响。催化剂性能受温度、催化剂用量以及反应时间影响,在80–120 ℃,催化剂活性随温度升高而升高,而CO2的活性随温度升高而降低[24]。文献中实验所用链转移剂种类、链转移剂与PO比例、催化剂用量均有差别,所以需综合考虑聚合产物分子量、产物选择性和碳酸酯单元摩尔含量等因素评价催化剂活性。在105 ℃条件下,以PEG1000为共配体的催化剂,产物中碳酸酯单元质量分数仅为9.4%,且分子量分布较宽。在80 ℃条件下反应,未加入共配体的催化剂产物中碳酸酯单元质量分数为58%,但PO转化率较低。而D2000作为共配体时,产物碳酸酯单元质量分数为25%,选择性为92%,分子量分布较窄。因此,与已报道的催化剂相比,在较温和反应条件下,DMC+D2000表现出更好的催化性能。
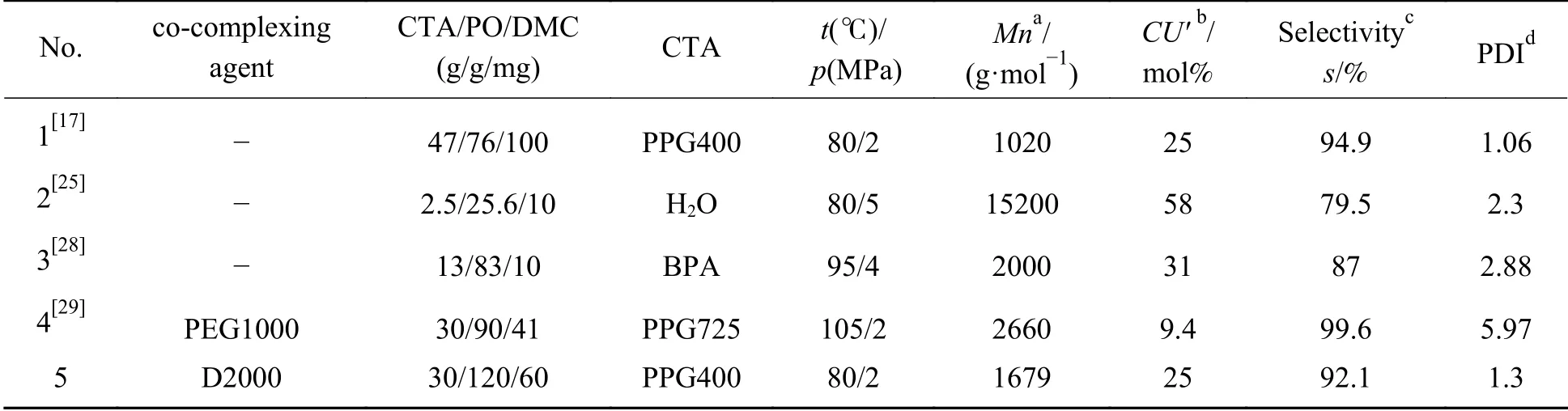
表4 不同催化剂催化CO2与 PO共聚反应对比Table 4 Comparison of different catalysts for the copolymerization of PO and CO2
2.5 产物结构表征
产物的1H NMR和GPC测试结果如图8所示。1H NMR (a)中,观察到聚醚碳酸酯二元醇中碳酸酯单元(δ=1.29–1.3)、聚醚链段(δ=1.14)以及环状碳酸酯(δ=1.5)的吸收峰,表明聚合物分子链中同时含有聚醚链段和聚碳酸酯链段,且两者是随机分布的。GPC(b)为反应产物的凝胶渗透色谱图,根据出峰时间确定为聚醚碳酸酯二元醇(t=8.64–9.46 min)和副产物环状碳酸酯(t=10.91 min)。与其他四种催化剂相比,DMC+D2000的产物出峰时间短,分子量大,分子量分布窄。
3 结 论
采用Span80、Tween80、D400和D2000四种含有不同结构、官能团的共配体对催化剂改性。
经Span80、Tween80改性后的催化剂结构没有发生改变,且催化剂粒径增加。由于自身的物理性质使催化剂颗粒形貌由“花朵”变为片层结构,不利于提高催化剂活性;经聚醚胺改性后的催化剂结构、结晶度发生改变,催化剂粒径减小。推测是因为聚醚胺与催化剂发生化学相互作用而保留在催化剂体系中,有利于提高催化剂活性。
从催化剂活性来说DMC+S80、DMC+D400、DMC、DMC+T80和DMC+D2000催化剂活性依次增加。其中,采用连续进料法在80 ℃,2.0 MPa,反应12 h的条件下,DMC+D2000催化剂的催化活性为1547 g/gcat.,聚合产物中碳酸酯单元占聚合部分的质量分数为25.3%,选择性为92.1%。
猜你喜欢
科学大众(2023年17期)2023-10-26 07:38:56
氯碱工业(2022年5期)2022-11-25 15:44:27
小天使·二年级语数英综合(2021年5期)2021-07-11 10:58:35
中国氯碱(2021年10期)2021-04-13 15:56:36
世界农药(2019年2期)2019-07-13 05:55:12
中学化学(2017年2期)2017-04-01 08:51:54
化工生产与技术(2016年5期)2016-03-13 10:07:26
股市动态分析(2015年12期)2015-09-10 13:18:31
石油化工技术与经济(2014年5期)2014-04-06 01:05:12
火炸药学报(2014年1期)2014-03-20 13:17:24
