
离子色谱-电化学法测定阿奇霉素中盐酸羟胺
2022-09-06 03:39徐艳梅韩彬郝丽娟高燕霞张素平
化学分析计量 2022年8期
徐艳梅,韩彬,郝丽娟,高燕霞,张素平
(河北省药品医疗器械检验研究院,石家庄 050200)
阿奇霉素为十五环含氮大环内酯类抗生素,是新型红霉素的最具代表的药物之一[1-2],由克罗地亚的普利瓦(Pliva)制药公司在20 世纪70 年代末开发[3-5],合成路线是以红霉素A 原料,经过肟化、贝克曼重排、还原和甲基化反应得到最终产物[6]。
盐酸羟胺是合成关键中间体——红霉素A 肟的主要原料,未反应完全的盐酸羟胺有引入药物的可能,属于潜在杂质。已报道盐酸羟胺结构的物质具有遗传毒性和致基因突变作用[7-9],致突变机理是改变DNA 的胞嘧啶结构,导致DNA 复制时碱基配对错误而诱发基因突变。依据欧洲共同体药物评审委员会(EMEA)、人用药品委员会(CHMP)《遗传毒性杂质限度指导原则》和人用药品注册技术要求国际协调会(ICH)的指导原则M7 《评估和控制药物中DNA 反应的(诱变的)杂质以限制潜在的致癌风险》与M7(R1)《评估和控制药物中DNA 反应性(致突变)杂质以限制潜在致癌风险》的相关要求,应对阿奇霉素原料中残留的盐酸羟胺进行测定,依据《化学药物质量标准建立的规范化过程技术指导原则》(CPH1-1)和《化学药物杂质研究的技术指导原则》(CPH3-1)中描述:“与已知毒性杂质结构相似的杂质,亦被认为是毒性杂质”的规定,因此本方法仍然按照毒性物质日允许暴露量为2 μg/d 限度进行控制。
然而盐酸羟胺在紫外光区无特征吸收,较难进行测定。目前常用的检测技术主要有借助氧化还原反应原理的滴定法[10]及复杂衍生化操作的间接分光光度法[11-14]等,其专属性不强,易受其它类似化合物干扰——尤其是具有相近性质的杂环仲胺盐,同时精密度和灵敏度较差,不适用于痕量羟胺的检测。离子色谱法近年来在分析化学领域的应用已日趋成熟,特别是在针对强极性、水溶性好、可电离的有机离子化合物分析方面表现出了独特的优势[15-17]。笔者建立了一种操作简单、专属性强、灵敏度高、重复性好的离子色谱法,用于阿奇霉素中痕量盐酸羟胺残留的检测。
1 实验部分
1.1 主要仪器与试剂
离子色谱仪:DIONEX ICS-5000 型,配备安培检测器(ED)和双泵系统(淋洗液单泵和柱后衍生AXP 泵),美国赛默飞世尔科技公司。
电子天平:XS205 型,感量为0.01 mg,梅特勒-托利多科技(瑞士)有限公司。
阿奇霉素原料药样品:批号分别为A20190503、A20190504、A20190505。
盐酸羟胺对照品:批号为100496-200801,纯度(质量分数)为100.0%,中国食品药品检定研究院。
甲基磺酸(MSA):纯度(质量分数)为99.5%,上海阿拉丁生化科技股份有限公司。
氢氧化钠溶液:质量分数为50%,美国赛默飞世尔科技公司。
实验用水为超纯水,电阻率为18.2 MΩ·cm。
1.2 溶液配制
盐酸羟胺对照品储备液:20 μg/mL,精密称取盐酸羟胺109.21 mg,置于50 mL 容量瓶中,用10 mmol/L MSA 溶解并定容,精密量取1 mL,置于50 mL 容量瓶中,用10 mmol/L MSA 稀释至标线,摇匀。
盐酸羟胺对照品溶液:0.2 μg/mL,取盐酸羟胺对照品储备液0.5 mL,置于50 mL 容量瓶中,用水稀释至标线,摇匀。
阿奇霉素样品溶液:取阿奇霉素原料药0.25 g,精密称定,精密加入5 mL 水,振摇30 s,过0.22 μm滤膜。
阿奇霉素样品加标溶液:取阿奇霉素样品约0.25 g,精密称定,置于10 mL 容量瓶中,用0.2 μg/mL 盐酸羟胺对照品溶液溶解并稀释至标线,摇匀。
MSA 溶液:30 mmol/L,精密量取MSA 0.967 mL,精密加入500 mL 水。
1.3 色谱条件
色谱柱:IonPac CS16 阳离子交换分析柱(250 mm×4 mm,美国赛默飞世尔科技有限公司),IonPac CG16 阳离子交换保护柱(50 mm×4 mm,美国赛默飞世尔科技有限公司);柱温:35 ℃;淋洗液:30 mmol/L MSA 溶液,等度洗脱,流量为0.8 mL/min;进样体积:25 μL;衍生试剂:500 mmol/L NaOH 溶液,流量为0.2 mL/min;检测器:电化学检测器,电极材料为金电极,参比电极为pH-Ag/AgCl电极;检测器温度:30 ℃;电位波形:积分脉冲安培检测,6 电位波形,安培电位波形表见表1。

表1 安培电位波形表
2 结果与讨论
2.1 淋洗液浓度的选择
在离子色谱分析过程中,淋洗液的组成、浓度及流量等都对出峰情况具有较大影响。其中,淋洗液的浓度对离子的保留时间影响较大,淋洗液浓度越大,保留时间越短,因此需要对淋洗液浓度进行考察,以选择较为合适的浓度。笔者对不同淋洗液浓度进行了考察,以色谱峰形、理论塔板数为指标,考察了淋洗液浓度对检测结果的影响。试验结果表明,当淋洗液浓度为20 mmol/L 时,色谱峰有较明显前延;当淋洗液浓度为30 mmol/L 和40 mmol/L 时峰形良好,故选择淋洗液浓度为30 mmol/L。
2.2 淋洗液流量的选择
淋洗液流量对于离子的保留时间也有较大影响,流量增大可缩短分析时间,但亦会影响色谱峰形,因此合适的淋洗液流量是进行分析不可缺少的前提。试验考察了淋洗液流量分别为0.7、0.8、0.9 mL/min 时的色谱峰形。结果表明,当流量为0.8 mL/min 时色谱峰形较好,因此选择淋洗液流量为0.8 mL/min。
2.3 柱温的选择
柱温对于色谱峰保留时间具有显著影响,不同离子在柱温变化时呈现出不同的变化趋势。为得到较好的色谱峰形及合适的保留时间,对柱温进行了试验考察,以色谱峰形、理论塔板数为指标,分别考察比较了柱温为25、35、45 ℃时的检测结果,表明盐酸羟胺的测定受柱温影响不大,故选择更易实现的35 ℃作为柱温。
2.4 专属性
以实验用水作为空白溶液,在优化的色谱条件下,分别分析空白溶液、盐酸羟胺对照品溶液及阿奇霉素品加标样品溶液,记录色谱图,结果如图1~图3。由图1~图3 可知,盐酸羟胺色谱峰峰形较好,且样品分析无干扰。

图1 空白溶液色谱图
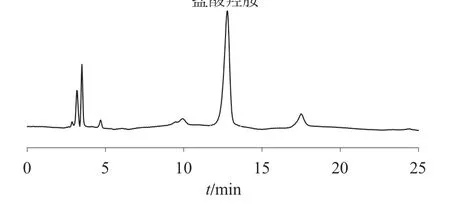
图2 阿奇霉素加标样品溶液色谱图
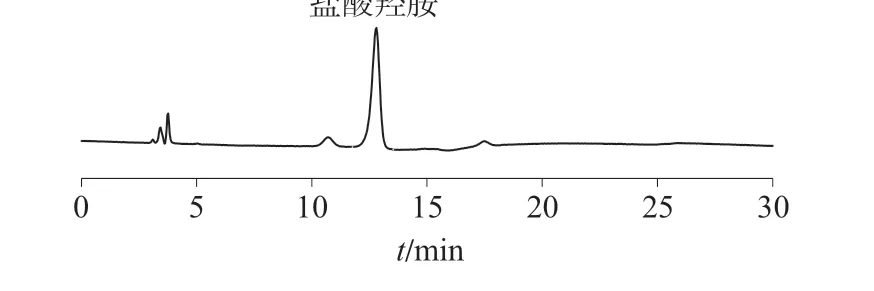
图3 盐酸羟胺对照品溶液的色谱图
2.5 线性关系
分别精密量取盐酸羟胺对照品储备溶液0.01、0.1、0.5、1.0、3.0、5.0、10.0 mL,分别置于7 只50 mL容量瓶中,用水稀释至标线,配制成盐酸羟胺质量浓度分别为0.004、0.04、0.2、0.4、1.2、2.0、4.0 μg/mL的系列标准工作溶液。精密量取上述溶液各25 μL,分别注入离子色谱仪,记录色谱图,以色谱峰面积为纵坐标,以盐酸羟胺质量浓度ρ(μg/mL)为横坐标进行线性回归,得到回归方程:y=28.523 7ρ-0.366 6,相关系数(r)为0.999 9。结果表明,盐酸羟胺在0.004 2~4.2 μg/mL 范围内线性关系良好。
2.6 定量限与检出限
精密量取阿奇霉素对照品溶液逐级稀释并测定,直至信噪比约10,即为定量限溶液。精密量取定量限溶液3 mL,置于10 mL 容量瓶中,加水稀释至标线,摇匀,即为检出限溶液。分别取定量限溶液和检出限溶液注入离子色谱仪,连续测定6 次,记录色谱图。结果表明,羟胺的定量限和检出限分别为0.207 ng/mL 和0.062 ng/mL。
2.7 加标回收与精密度试验
精密称取阿奇霉素原料药0.25 g,共9 份,置于5 mL 容量瓶中,分别精密加入盐酸羟胺对照品储备溶液25、50、75 μL 各3 份,加水振摇30 s,用水定容至标线,过滤。精密量取上述溶液注入离子色谱仪,记录色谱图,测得量与加入量的比值即为回收率,结果列于表2。由表2 可知,盐酸羟胺的回收率为94.50%~100.46%,相对标准偏差为1.9%。说明本方法符合验证要求,准确度良好。
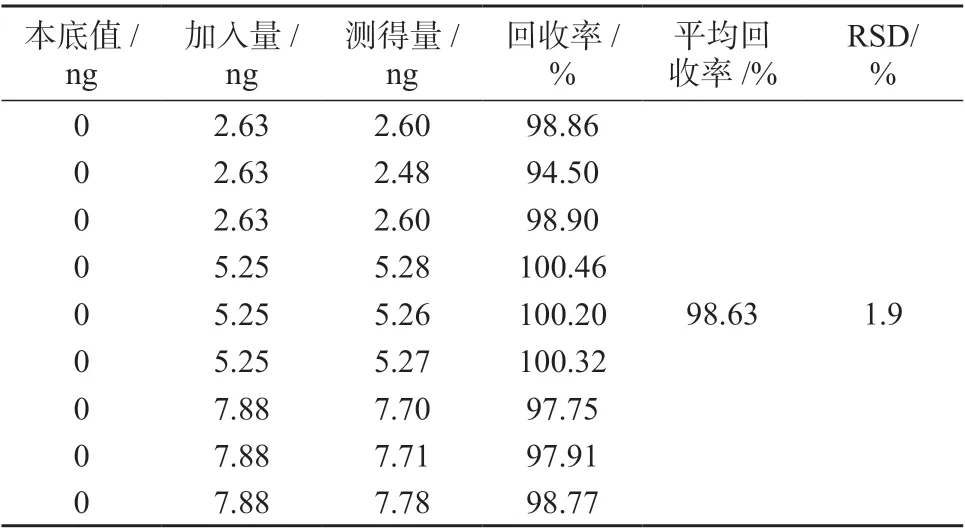
表2 加标回收与精密度试验结果
2.8 稳定性试验
取新鲜配制的盐酸羟胺对照品溶液和阿奇霉素加标溶液,于室温放置0、1、2、3、4 h,精密量取不同放置时间的上述溶液注入离子色谱仪,记录色谱图,色谱峰面积测定结果见表3。由表3 可知,对照品溶液及阿奇霉素加标样品溶液放置4 h 时盐酸羟胺色谱峰面积分别降低10.4%及10.1%左右,这提示盐酸羟胺在水溶液中不稳定,样品溶液宜临用新制。
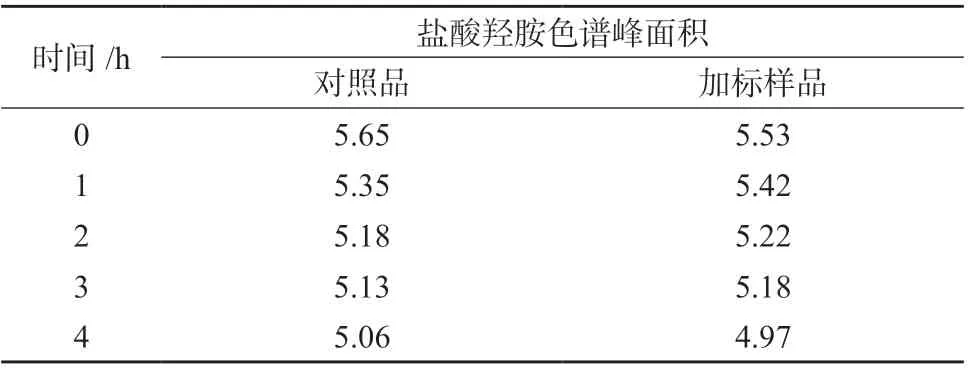
表3 对照品与样品溶液稳定性试验
2.9 强制破坏试验
取阿奇霉素原料药样品,分别在酸、碱、加热、光照和氧化条件下,按照表4 方法进行强制降解试验,按照1.2 项下方法制备溶液,所得溶液注入离子色谱仪,记录色谱图,色谱图如图4~图8 所示。由图4~图8 可知,阿奇霉素降解产物对盐酸羟胺的测定无干扰。

表4 破坏实验方法

图5 碱破坏试验色谱图
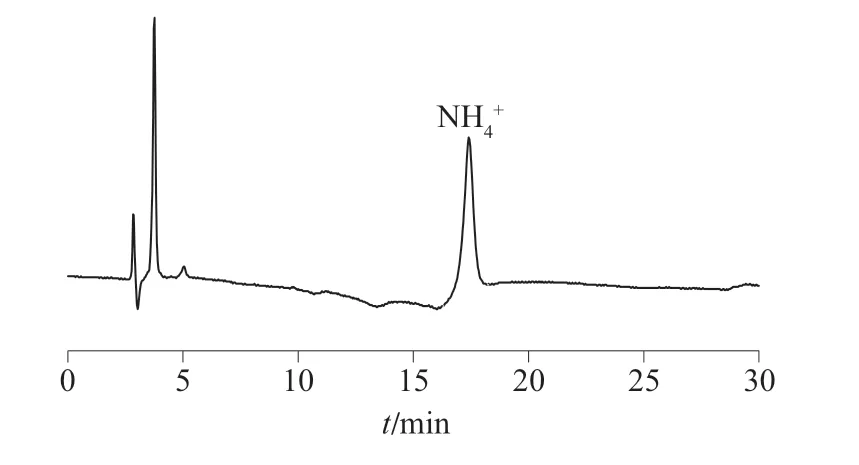
图6 热破坏试验色谱图
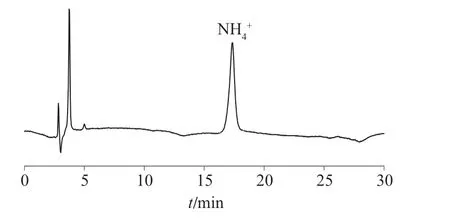
图7 光破坏试验色谱图

图8 氧化破坏试验色谱图
3 结语
建立了离子色谱-电化学法测定阿奇霉素中残留的盐酸羟胺的方法,通过调节淋洗液浓度及流量,获得了最佳淋洗液条件,可对盐酸羟胺进行准确定量。该法具有良好的线性、准确性和重现性,检出限低。本方法具有较好的实用价值,对测定药物中残留的盐酸羟胺具有一定的借鉴作用。
猜你喜欢
医学前沿(2021年18期)2021-04-14
中国应急管理科学(2021年4期)2021-04-13
四川蚕业(2020年4期)2020-02-10
中学生数理化·高一版(2020年9期)2020-01-02
中学化学(2019年4期)2019-08-06
学苑创造·A版(2019年4期)2019-05-10
考试周刊(2018年68期)2018-09-17
恋爱婚姻家庭·养生版(2013年6期)2013-05-14
祝您健康(1986年4期)1986-12-30
