
注意力特征融合的蛋白质-药物相互作用预测
2022-09-06 07:30李金星冯振华宋晓宁於东军
计算机研究与发展 2022年9期
华 阳 李金星 冯振华 宋晓宁 孙 俊 於东军
1(江南大学人工智能与计算机学院 江苏无锡 214100)
2(萨里大学计算机系 英国吉尔福德 GU2 7XH)
3(南京理工大学计算机科学与工程学院 南京 210094)
(7211905018@stu.jiangnan.edu.cn)
预测蛋白质-药物相互作用是早期药物筛选中的关键步骤.据美国药物研究与制造商协会调查,新药研究成本[1]占用整个制药业收益的75%.此外,仅有不到5%的经初筛命中的化合物可用于临床实验,传统的筛选方法更是要消耗2~3年的时间,极大程度地耗费了研究人员的精力和时间.借助计算机进行虚拟筛选[2]药物花费时间短、准确性高,有效降低了该任务的成本.而该方案的核心是依靠计算机预测出蛋白质和药物之间的相互作用(protein-drug interaction, PDI)进行药物筛选[3].
预测蛋白质-药物的相互作用主要包含3个步骤:1)对药物分子式进行量化并提取其特征;2)对蛋白序列进行量化并提取其特征;3)选择合适的分类模型,预测药物和蛋白质是否存在相互作用[4].不难看出,与其他模式识别任务类似,有效提取特征的方法是进行蛋白质-药物相互作用预测的关键.
在提取药物特征方面,药物分子量化的理论来源于定量结构与活性关系(quantitative structure activity relationship, QSAR)[5].该关系源自传统构效关系,并在此基础上与化学中常见的经验方程相结合,在药物化学领域具有广泛且深远的影响.该方法把人们对构效关系的认知从定性水平上升到定量水平.从其实际影响来看,定量结构与活性关系揭示了药物分子与生物大分子结合[6]的模式,指示化合物的某些生物活性可以通过数学模型量化其分子结构特征[7]来获得,并给出了量化特征的理论依据.在蛋白质特征提取方面,蛋白序列的量化方式[8]主要来源于对氨基酸残基的特征[9]嵌入.当下主流的做法有自相关矩阵嵌入和残基序列[10]结合嵌入等.
除了蛋白质和药物的特征提取方式,分类模型的选择[11]和设计也是预测蛋白质和药物相互作用的重要研究内容.现有的预测算法主要分为两大类:传统机器学习[12]方法和深度神经网络[13]方法.传统机器学习方法包括支撑向量机(support vector machine, SVM)[14]、随机森林(random forests. RF)[15]、K最近邻(k-nearest neighbor, KNN)[16]分类算法以及逻辑回归模型等.基于深度神经网络的方法主要包括长短期记忆网络(long short-term memory, LSTM)[17]、卷积神经网络(convolutional neural networks, CNN)[18]和图神经网络(graph neural network, GNN)[19]等.在现有算法中,大多方法都根据靶蛋白种类划分成4类去解决,即分别在酶、离子通道、G蛋白耦联受体和核受体蛋白中进行预测.这么做的主要原因是这4类蛋白的类间差异过大,混在一起训练会使模型变得很难收敛.因此,这些方法训练出的模型可适用面较窄,往往只局限于预测某一类蛋白和药物的相互作用.当无法判断一种新的未知蛋白的真实属性时,该类方法的鲁棒性和实用价值会大大降低.
为解决上述模型泛化能力不足的问题,由Lee等人[20]提出的DeepConv-DTI模型对变长的蛋白序列进行补零定长,将差异较大的蛋白序列特征固定在相同维度空间中,并采用卷积神经网络提取蛋白质的低维实值特征,有效拓宽了模型的泛化性.与之相似的是,Öztürk等人[21]提出的DeepDTA模型还额外添加配体最大公共结构和蛋白质结构域特征来提升模型的效能.然而,这2个模型提取药物特征的方法仍然存有不足,仅使用扩展连通性指纹Morgan[22]作为量化药物的方法无法提取分子的结构信息.Morgan指纹编译是一种圆形拓扑指纹,先枚举药物分子中所有可能的原子位次,然后将各原子周边的原子信息增加到该原子上形成数组,并散列至一个数字,最后通过计算连通性选择最合适的位次得到对应的分子指纹.这种方法可有效提供分子特征的细节,但药物分子结构信息的缺乏导致蛋白质-药物相互作用预测的性能明显不足.而CPI-GNN[23]和GraphDTA[24]等模型借助图卷积网络提取药物特征一定程度上获取了药物分子的结构特征,在其实验中取得相对不错的效果.但是图卷积模型对分子结构的辨识度会随着药物种类的增多而下降,也会致使模型的整体性能下降.
此外,文献[20-21,23-24]所提的方法大都使用多层感知器(MLP)模型[25]预测蛋白质-药物的相互作用.该方法无法凸显药物特征中重要的局部信息,致使整个模型的预测性能达不到最佳效果.为解决这一问题,Wang等人[26]在2020年初提出了一种使用深度长短期记忆网络(deep long short-term memory, DeepLSTM)的方法预测蛋白质-药物相互作用,在酶和G蛋白耦联受体的作用预测中都取得了最佳效果.虽然这一方法无法有效预测大规模数据下的PDI,但论述了通过添加时序信息可有效捕捉更多的鉴别PDI的特征.2020年Chen等人[27]提出基于自然语言处理(natural language processing, NLP)中Transformer骨干网络[28]中的TransformerCPI模型.该模型利用自注意力机制有效地捕捉化合物和蛋白质之间的关联特性,在各公开数据集中均表现良好.但因为其提取的蛋白特征不够充分导致其在Kinase数据集上的收敛效果不够理想.
为进一步提升在大规模数据下PDI实验的预测效果,本文结合前人的工作提出了一种将深度卷积神经网络与自注意力循环网络相结合的模型.在药物特征提取方面,由于Morgan无法提取药物分子的子结构特征,本文在其基础上增加了Mol2Vec向量[29]嵌入.Mol2Vec是一种无监督的机器学习方法,用于学习分子亚结构[30]的向量表示.与Word2Vec模型[31]一致,密切相关单词的向量在向量空间中非常接近,Mol2Vec可以学习指向与化学相关子结构方向相似的分子子结构的向量表示,并以对各个子结构做向量求和的方式来编码化合物.Mol2Vec模型克服了常见复合特征表示的缺点,如稀疏性和位冲突,在多个化合物特性和生物活性数据集上验证了其预测能力.为了补充更多药物特征信息,本文在原基础上额外添加由消息传递神经网络(message passing neural networks, MPNNs)[32]提供的图特征,经实验验证在数据规模较小的实验中添加该特征可有效提升模型的性能,但对数据规模较大的实验模型效果不明显.据此,本文先用循环网络采集Morgan指纹和Mol2Vec向量的重要信息并将两者进行融合,再根据数据规模差异考虑是否融合图特征,最后本文用密集型卷积网络进一步提取出药物分子的特征.
在蛋白特征提取方面,本文根据生物活性对蛋白质的氨基酸序列进行归类[33],这样可有效减少蛋白嵌入特征的稀疏性.并借鉴了Lee等人[20]的经验,对变长的蛋白序列编码进行补零定长并嵌入至等长的特征空间中.为了模型收敛得更好以及蛋白质特征采集得更详细,本文选择密集型卷积网络DenseNet[34]提取蛋白特征,该模型可直接连接来自不同层的蛋白特征,通过特征重用可有效提升效率和精度.接着利用药物特征对蛋白特征进行注意力针对训练,这样得到的蛋白特征除了包含蛋白序列本身信息之外还包含蛋白药物的关系信息.其结果与药物特征一同放入双方向门控循环单元提取PDI的特征信息.
门控循环单元[35]是循环神经网络(recurrent neural network, RNN)中的一种门控机制.与其他门控机制相似,门控循环单元旨在解决标准循环神经网络中的梯度消失(爆炸)问题,并同时保留序列的长期信息.由此得到的PDI特征信息再经过自注意力机制对重要区域进行加权,以及经过全连接层映射成长度为2的1维向量.最后放入Softmax激活函数中进行归一化处理作为蛋白质-药物相互作用的预测结果.除此之外,本文结合所提模型设计了可用于药学研究的界面,并提供了筛选抑制乙酰胆碱脂酶(AChE)和丁酰胆碱酯酶(BuChE)药物的方法.这类药物对治疗阿尔兹海默症具有重要意义[36],本文也借此进一步阐述了模型的实用价值.
综上所述,本文的主要创新点包括:
1) 提出一种复合提取药物特征的方法,将Morgan指纹编译、Mol2Vec向量以及MPNNs图结构特征合理融合,丰富了药物特征信息;
2) 根据生物活性对氨基酸进行归类,有效降低蛋白特征的稀疏性,采用密集型卷积网络提取特征,实现特征重用,提升模型的效率和精度;
3) 发现深度网络结合门控循环单元提取蛋白质和药物特征,结合能强化蛋白质和药物关系特征的注意力网络,可有效提升模型的性能.
1 本文方法
1.1 药物特征表示
药物特征提取是预测PDI的重要环节.其目的是提取药物的鉴别特征,使得分类器可以更好地理解药物性质并能区分出不同药物之间的差异.因此,有效的药物特征需要具备可鉴别性和典型性,且类间的距离相对较远而类内的距离相对较近.这样便于提升分类模型的预测精度.常用的药物特征嵌入方法有2种,包括Morgan指纹和Mol2Vec向量.
Morgan指纹又称扩展连通性指纹,是一种圆形指纹.借助Morgan指纹编码药物分子式如图1所示:
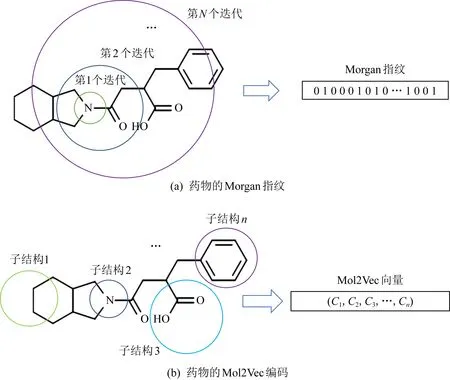
Fig. 1 Drug feature embedding图1 药物特征嵌入
药物特征嵌入的步骤为:首先根据给定的半径分析每个原子的环境和连通性;然后散列编码所有可能形成的结构;最后根据散列算法将编码信息放缩到预定长度.这种方法的代表性虽然很全面,但由于尺寸较大且信息过于离散,难以合理地表达药物的子结构信息.另一种常用的嵌入药物分子式特征的方法是Mol2Vec向量编码.Mol2Vec是从NLP中的Word2Vec演变而来,可以学习分子间化学性质指向相似的子结构信息.其编码方式为将各个子结构向量的和作为化合物的特征向量.该方式可将药物的子结构特征表示得很清晰,具有很强的典型性,也是对Morgan特征的一项重要补充.
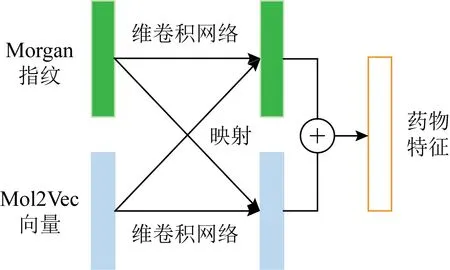
Fig. 2 The feature fusion module图2 特征融合模块
为了提取具备鉴别性和典型性的特征,本文将图1所示的2种药物特征嵌入方法结合使用.首先,通过双向门控循环单元对嵌入特征进行特征抽取;然后将2种方法所得药物特征按照图2的方式融合;最后将融合特征放入密集型卷积网络特征抽取,作为与蛋白结合预测的药物特征.其中,融合方法为先用1维卷积(1×3)将原特征转至下层特征,随后使用映射矩阵分别将Morgan特征和Mol2Vec特征映射到与对方相同的特征空间中并与下层特征相加,2个映射矩阵互为转置,公式为:
FM1=Conv1d(FM0)+VFN0,
(1)
FN1=Conv1d(FN0)+VTFM0,
(2)
其中,FM0和FM1分别是Morgan原特征与下层特征,FN0和FN1分别是Mol2Vec原特征与下层特征,V是可训练的映射矩阵,其维度分别是FM0和FN0的长度.使用同映射矩阵可以使2类特征的关联性更强、模型更容易收敛,最后将两者下层特征相加得到融合特征.
同时,本文发现消息传递网络可以抽取药物的平面结构特征,存有提升实验效果的可能.MPNNs由Google的研究人员提出并用于预测量子化学性质,可有效应用于小样本模型[37].具体算法为:首先,构造初始状态集,每个状态用于图中的每个节点;然后,使每个节点与其邻居交换信息以进行消息传递,这样每个节点状态将包含对其直接邻居的感知.重复这2个步骤,每个节点便可获得其2阶邻域的信息,依此类推.达到预想次数的“消息回合”,便可将所有上下文的节点的状态转换表征为整个图的特征.其节点更新权重的公式为
(3)
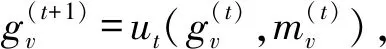
(4)
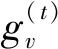
如图3所示,本文尝试采用特征融合模块将MPNNs模块所提取的药物图特征与由Morgan和Mol2Vec融合的特征相融,然而实验效果在不同规模数据集上的表现有所差异,具体的内容将3.2节进行详细的论述.实验表明,MPNNs提供的图结构特征会让模型在小规模数据集上表现得更好,但对数据规模较大的实验效果不明显.
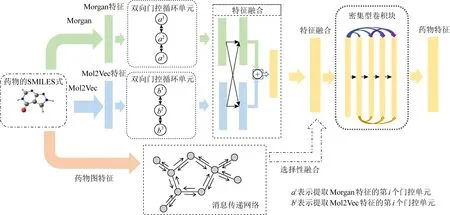
Fig. 3 The drug feature extraction model图3 药物特征提取模型
1.2 蛋白质特征表示
本文沿用Lee等人[20]的方法对蛋白质一级结构氨基酸残基序列进行编码,将字符形的序列嵌入到离散的整形向量中.本文对蛋白质序列做了一步预处理,在本文所提各数据集中蛋白质序列是由22类氨基酸混合组成,若以此作为文本,3个氨基酸片段作为词划分蛋白,便有22×22×22=10 648种组合,这便导致蛋白的特征矩阵过于稀疏.为解决这一问题,本文借用Che等人[33]使用的方案,根据生物化学的特性将22种氨基酸归类为6种:a={H,R,K},b={D,E,N,Q},c={C,X},d={S,T,P,A,G,U},e={M,I,L,V},f={F,Y,W}.
这样序列MSPLNQSAEGLPQEASNRSLN便可以转化为eddebbddbdedbbddbadeb,该方法得到组合数为6×6×6=216种可显著降低特征矩阵的维数.同时,本文使用DenseNet提取蛋白特征.DenseNet提出了一个非常激进的密集连接机制,即互相连接所有的层.具体来说就是每个层都会接受其前面所有层作为其额外的输入,每层都会与前面所有层在维度上连接在一起并作为下一层的输入.对于一个L层的网络,DenseNet共包含(L+1)L/2个连接.如图4所示,直接连接来自不同层的蛋白特征可有效提升实验的效率和精度.
本文采用1维卷积从上层模型提取特征,卷积提取特征的公式为

(5)
其中,函数x(t)和q(t)表示卷积的变量,p表示积分变量,t表示使函数q(t-p)位移的量,*表示卷积.蛋白质序列信息在经过特征嵌入、密集型卷积网络、最大池化、全连接后转变为128维的特征向量,与药物的特征向量一同放在分类模型中以预测PDI结果.
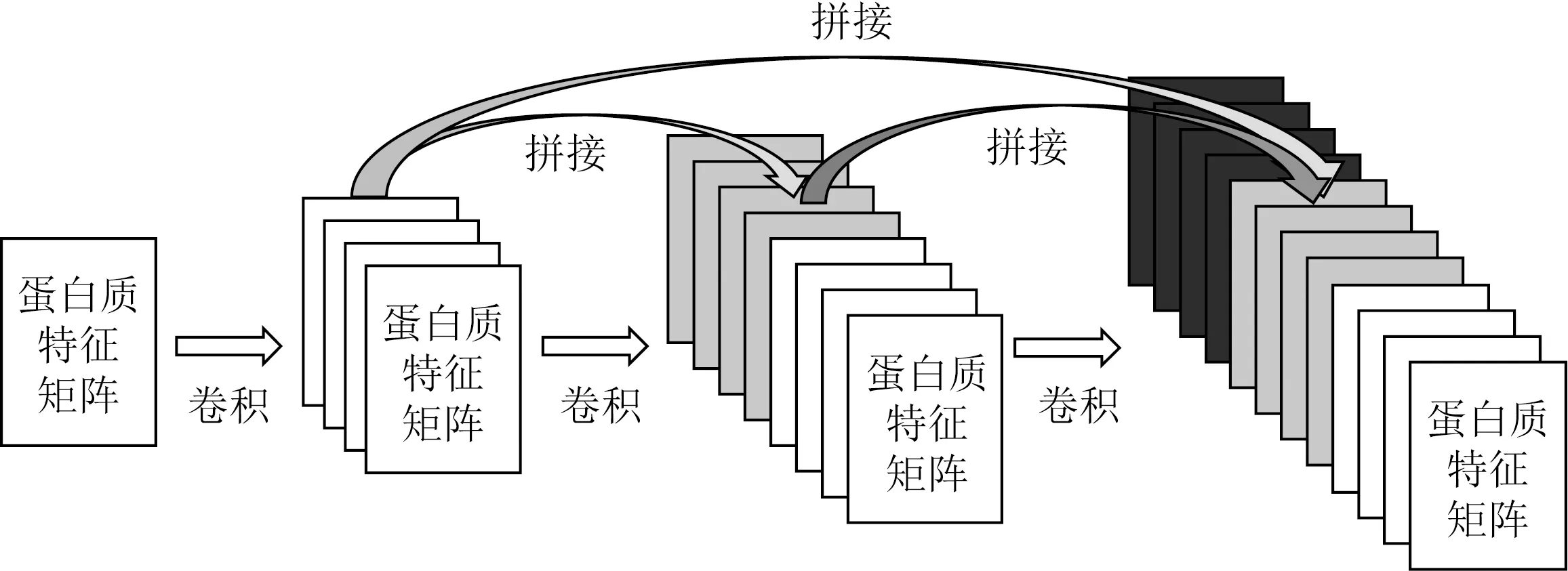
Fig. 4 The dense protein feature extraction model图4 密集型蛋白特征提取模型
1.3 模型整体框架
本文方法的整体框架如图5所示.该框架由3个部分组成:蛋白质、药物特征提取模块以及预测PDI模块.本文采用的是端到端的训练模型,即分类器与2个特征模型的训练任务同时进行.流程包括:

Fig. 5 The overall framework of the proposed method图5 本文方法框架
1) 药物分子经过2种特征嵌入后,由双向门控循环单元和邻域卷积先后抽取药物特征,并对由卷积模块所提取的蛋白特征做注意力权值增强.具体做法即给定一个药物分子特征向量Fdrug和蛋白质子序列特征向量P=(P1,P2,…,Pi),随后为其构造一个关于Fdrug的注意力矩阵.根据不同注意力权重来计算不同子序列对药物分子的重要性.公式为
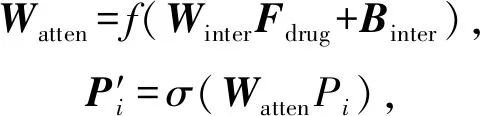
(6)
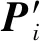
2) 拼接蛋白质特征和药物特征,并用自注意力机制[38]对PDI信息进行加权提取,具体做法即给定拼接后PDI特征向量cinter,训练一个自注意力矩阵Wself-atten对相互作用信息区域进行加权学习,公式为
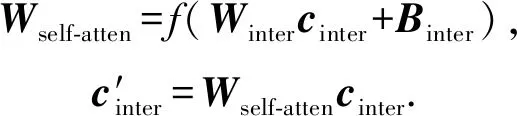
(7)
本文将在3.2节对2块注意力机制的使用效果进行验证.
1.4 损失函数和模型优化
本文使用交叉熵损失作为训练模型的损失函数:
(8)
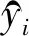
为了避免发生过拟合的情况,本文用L2范式作为惩罚项来约束模型的优化:
(9)
其中,W和b是模型全局的权重和偏置,λ是惩罚因子.同时本文嵌入dropout层[40]来辅助解决这个问题.为兼顾训练的效率和分类的精度,本文用Adam优化器[41]来更新模型的权值.
2 数据集
本文使用BindingDB,Kinase,Human,C.elegans这4个数据集来验证模型的效果.BindingDB是从大量科学文献中采集的可公开访问的蛋白质药物相互作用数据库.在2018年,由Gao等人[42]按照以下规则从该数据库中采集39 747个正样本和31 218个负样本制成用于评估PDI模型的BindingDB数据集.规则为:
1) 所记录的药物分子具有化学标识符(PubChem CID)以及以smiles表示的化学结构;
2) 所记录的蛋白质也需具有数据标识符(Uniprot ID)以及序列表示和基因本体注释;
3) 记录具有IC50值,即相互作用的主要指标;
4) 因为专注于小分子药物,所有的化学分子量均需小于1 000道尔顿;
5) 遵循Wang等人关于活动阈值的讨论,若IC50小于100 nm则记为正,IC50大于10 000 nm则记为负.
该数据集按如表1的方案来划分训练、验证和测试的子集,其中验证和测试的子集包含训练集中未被观察到配体或蛋白质的PDI样本.因此,BindingDB数据集可以评估模型对未知药物和蛋白质的鲁棒性.
Kinase数据集是由Chen等人[27]基于KIBA数据集构建而成,包含229个蛋白样本和1 644个药物样本.KIBA数据集已涉及多种用于测试活性的评价机制.对比多种生物活性评分,可有效减少由人为因素给数据集造成的偏差.同时Kinase的负样本远多于正样本,如表2所示.
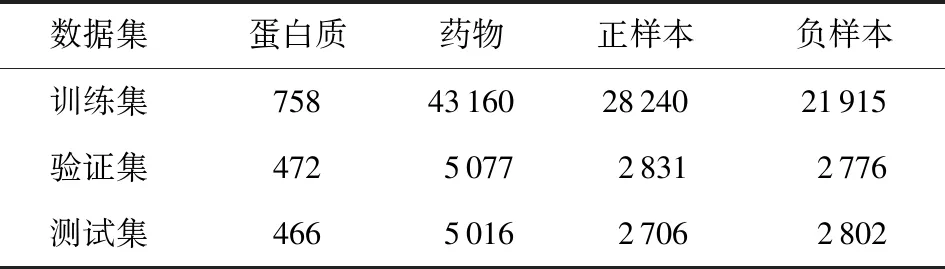
Table 1 The Distribution of BindingDB Dataset表1 BindingDB数据集分布

Table 2 The Distribution of Kinase Dataset表2 Kinase数据集分布
用此数据集可有效测试模型在样本不平衡下的性能,而在实际药物筛选过程中,没有相互作用的样本显然比有相互作用的样本多得多,因此模型在不平衡数据集下的表现也十分重要.
数据集Human和C.elegans是由Tsubaki等人[23]在2019年根据前人的工作汇总而成,其中Human数据集涵盖852种人类蛋白以及1 052种药物分子,数据共存有3 369个正样本和2 843个高度可信的负样本.C.elegans数据集包含2 504种线虫蛋白和1 434种药物分子,数据共存有4 000个正样本和3 511个高度可信的负样本.然而这2个数据集没有划分出训练和测试的子集,因此本文采用交叉验证的方式来评估模型在这2个数据集上的表现.本文所有数据及所提出的模型程序[43]均可从github上获取.
3 实验结果与讨论
3.1 实验环境以及参数设置
本文实验所配置的硬件:采用的中央处理器和显卡分别为Intel Core i7-8700k和NIVADA GeForce RTX 2060 s,并配用Windows10的操作系统.本文在Python3环境下的Keras深度学习框架上训练和评估模型,并使用Sklearn等机器学习工具处理实验数据.训练模型时,超参数的设置对模型优化有很大影响.本文优先固定参数优化的学习率,在此基础上使用网格搜索法对其他各参数进行寻优.多轮实验最终确定了模型的超参数设置,如表3所示:
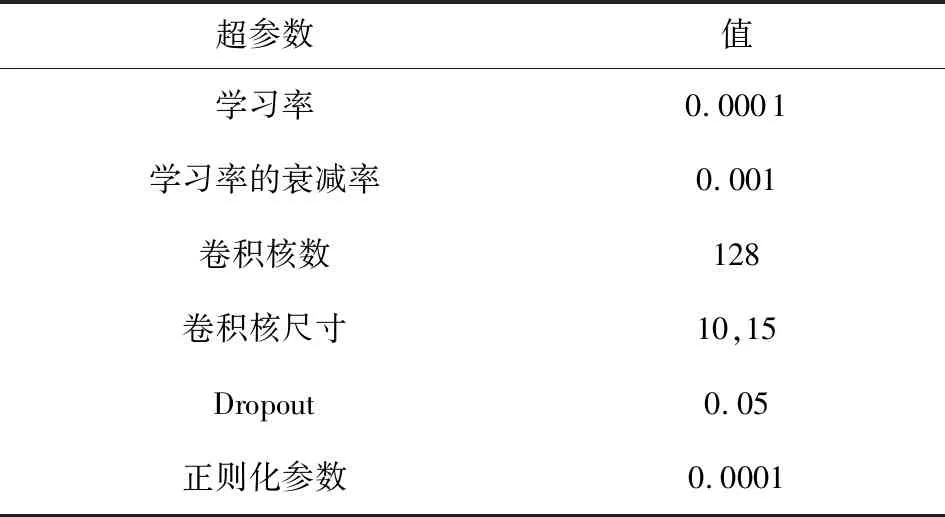
Table 3 The Hyper-Parameter Settings表3 超参数设置
本文使用2个指标评估各模型:
1) ROC曲线下的面积AUC.ROC曲线上的每个点是由2个指标的值来确定坐标,即真阳率(true positive rate,TPR)和假阳率(false positive rate,FPR),TPR=TP/(TP+FN),FPR=FP/(TN+FP).其中TP表示阳例预测为正例子的个数,FP表示阴例预测为正例子的个数,TN表示阴例预测为负例子的个数,FN表示阳例预测为负例子的个数.
2) PR曲线下的面积AUPR.PR曲线上的每个点是由2个指标的值来确定坐标,即精准率(precision,P)和召回率(recall,R),P=TP/(TP+FP),R=TP/(TP+FN).
3.2 消融实验
本文仿照DeepconvDTI的方法提取药物特征,即药物的表征由Morgan向量构成.结合本文所提模型在BindingDB数据集上的训练,在测试集上的最优结果的AUC=0.954.经调研发现,仅用Morgan向量表征药物易于忽视分子的亚结构信息,而这部分内容对量化药物特征信息十分重要.因此,本文使用涵盖药物亚结构信息的Mol2Vec向量对Morgan向量进行补充,并用DenseNet多尺度提取分子特征进一步优化了模型,最优结果AUC=0.963.该结果证实了药物特征提取方法的优化对模型整体预测能力的提升具有积极意义.
Riba等人[37]在研究图网络主动学习分子隐式结构特征的过程中表明:消息传递神经网络(message passing neural networks,MPNNs)可以帮助模型学习分子的结构信息.在此基础上,本文试图添加分子平面结构图的特征来进一步优化模型,并在原框架基础上添加MPNNs模块,然而在BindingDB数据集上测试效果相比原先模型下降了0.5%,这并不符合本文的预期.经仔细查阅发现,在Riba等人的工作中,数据的规模相对较小,于是本文在Human和C.elegans数据集上做了相同的消融实验,结果如表4所示:
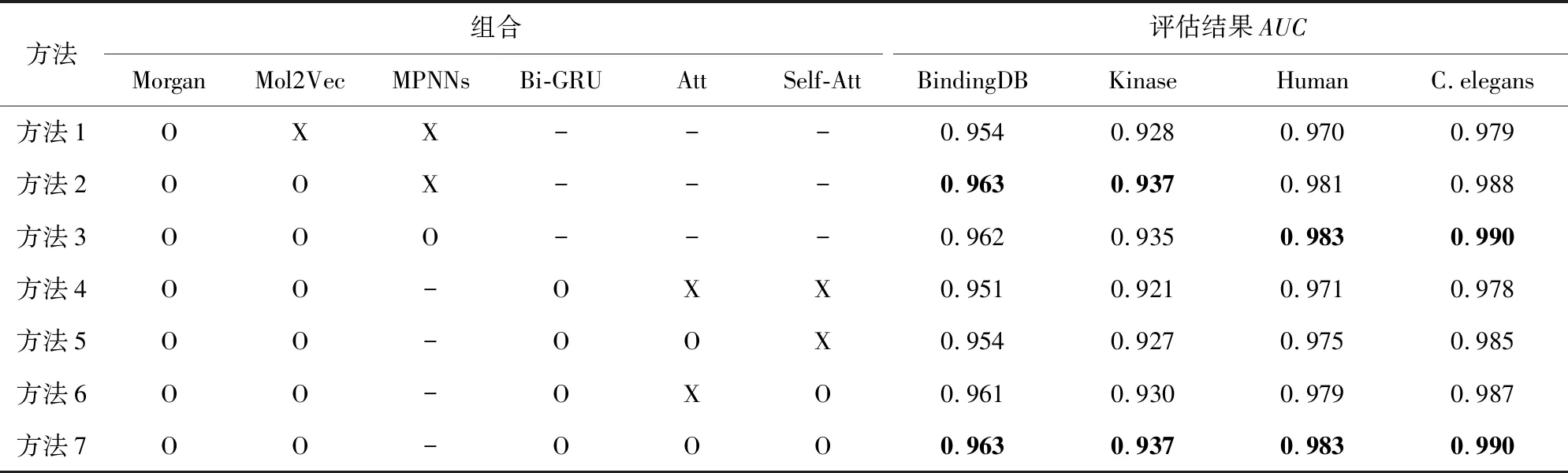
Table 4 The Ablation Study Based on Four Datasets表4 基于4个数据集上的消融实验
实验发现,添加MPNNs模型,会使模型整体在Human数据集和C.elegans数据集上提升0.2%,而在规模相对较大的BindingDB数据集和Kinase数据集上的效果分别下降了0.1%和0.2%.对此本文根据训练周期(epoch)的更迭,在BindingDB和Human数据集上绘制了不同药物特征组合下的样本类内距离和类间距离的变化趋势图.样本类内距离和类间距离计算公式为
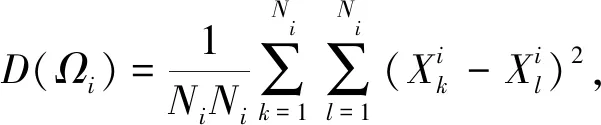
(10)
(11)
根据图6所示,在数据规模相对较大的BindingDB数据集中,仅使用Morgan指纹和Mol2Vec向量结合量化特征,会让样本随着epoch的迭代类间距变得更远、类内距离变得更近,这为分类器的判别提供了很大的便利.而添加MPNNs模型后在BindingDB数据集上的效果略微下降,但在Human数据集上的效果却有所提升,也从侧面论证了MPNNs的介入有益于模型在中小规模数据下的收敛,但对数据规模较大的实验效果不明显.
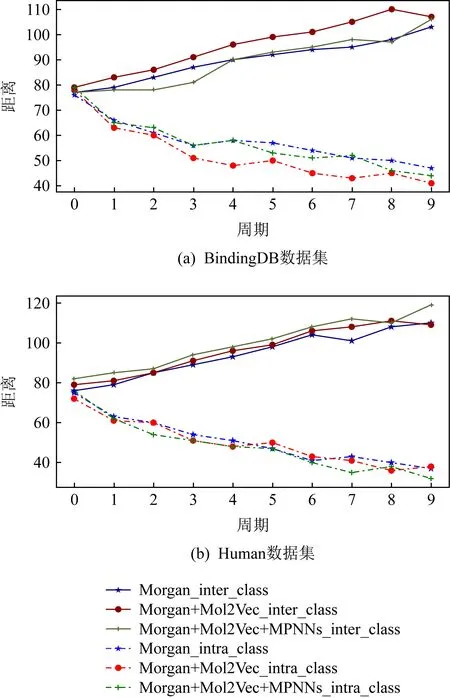
Fig. 6 The trend of intra-class distances and inter-class distances图6 类内距离和类间距离趋势图
确定了提取药物特征的模块后,据多轮实验结果发现,使用双方向门控循环单元做分类模块的性能最佳.在此基础上,本文开始研究如何利用注意力模块增强模型对重要信息的感知.如表4的方法4~7所示,模型在BindingDB测试集上的初始AUC=0.951.本文试图增强蛋白特征与对应药物特征的关联性,便在两者之间添加注意力模块,其结果相比初始AUC提升了0.3%.在此基础上,本文通过自注意力模块对合并的特征向量进行重要信息加权将模型的性能提升到最佳,其测试的AUC=0.963.除此之外,本文也附加仅添加自注意力模块的实验和在其他数据集上的测试结果,发现在各数据集上自注意力模块都可以使模型的精度提升近1%.
3.3 对比实验
本文用4个数据集检验所提模型,并与前人所提的同类模型进行对比实验,其中包括Tsubaki工作中提到的最近邻模型(KNN)、随机森林(RF)、L2逻辑回归、支持向量机以及CPI-GNN模型,不过因为在文献[23]中未涉及除了CPI-GNN以外的各个模型的参数细节,所以除了Human和C.elegans数据集外本文不再讨论前4类模型的性能.此外,本文还添加由Nguyen等人[24]提出的GraphDTA模型、由Lee等人[20]提出的DeepConvDTI模型以及由Chen等人[27]提出的GCN模型和TransformerCPI模型的讨论.值得注意的是,这些模型均是近年来PDI预测的典型模型.
如表5所示,本文依次在BindingDB数据集上比较提到的GraphDTA,GCN,CPI-GNN,Trans-formerCPI,DeepConvDTI和本文所提模型,前4个模型结果均从文献[27]中获取,DeepConvDTI模型与本文所提模型的结果都是在实验中经过调整参数所得到的最优结果.从表5可清晰地发现,本文所提模型的预测结果比当前前沿模型更佳;AUC较基线水准提升了0.019,与TransformerCPI相比提升了0.012.
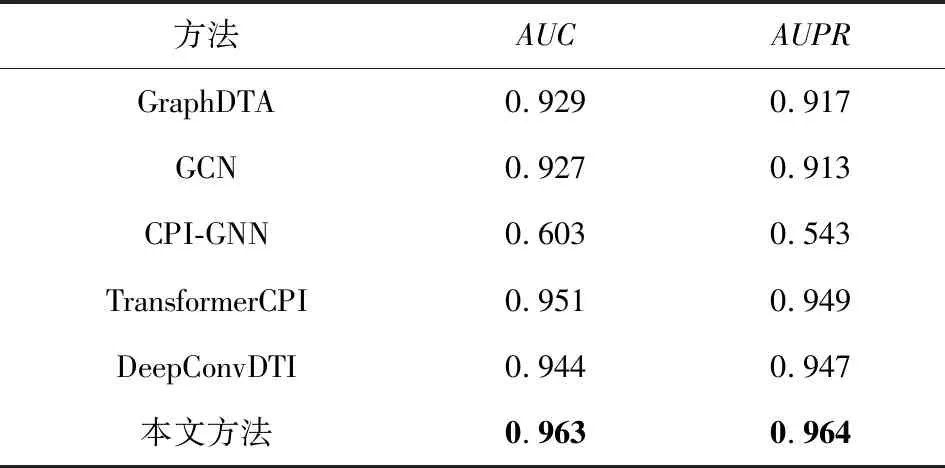
Table 5 Comparative Experiments on BindingDB Dataset表5 BindingDB数据集上的对比实验
在实际应用中,蛋白质和药物相互作用的负例个数远超正例个数,因此控制负例样本远多于正例样本更贴合实际情形.为验证模型在这一情形上的性能,本文在Kinase数据集上与文献[27]所涉及的各个模型进行对比.在第2节详细介绍了Kinase数据集因样本不平衡问题导致很多模型在此数据集上很难收敛.而从表6可以发现,与另外4个模型相比,本文方法在仿真的不平衡数据集上的表现依旧优异,具备突出的实际应用价值.
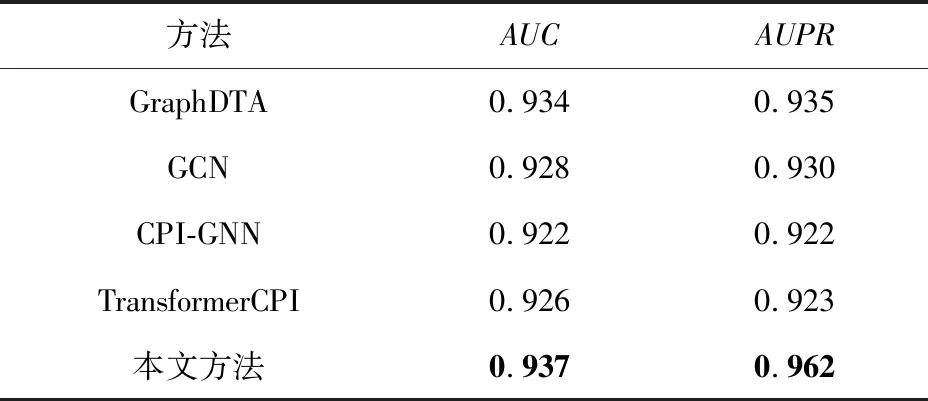
Table 6 Comparative Experiments on Kinase Dataset表6 Kinase数据集上的对比实验
最后,本文再次在广泛使用的Human和C.elegans数据集上评估新模型的性能.由于这2个数据集未划分出用于训练和测试的子集,本文采用交叉验证的方式评估各个模型.为了证明实验结果具备比较价值,本文划分数据的方式与前人相同,即按4∶1的比例划分训练集和测试集,评估体系也与Tsubaki等人[23]保持一致.本文将数据次序打乱随机划分了10次,实验结果的均值和方差分别如表7和表8所示.结果足以证明,本文方法在精确度以及稳定性上相比其他同类模型都更为优异.
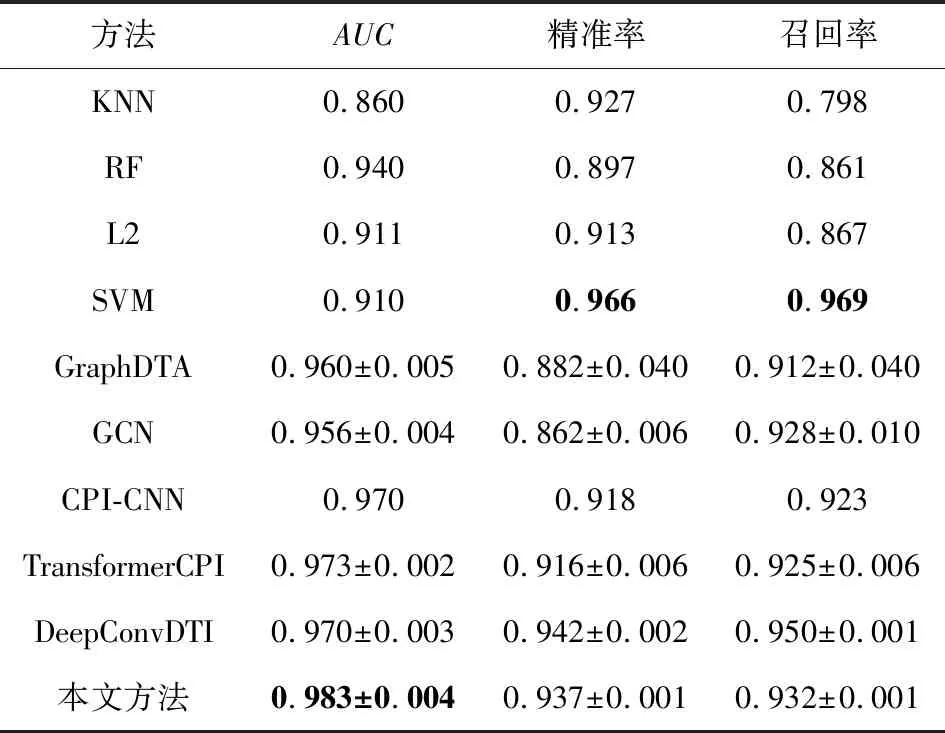
Table 7 Comparative Experiments on Human Dataset表7 Human数据集上的对比实验
因为C.elegans数据集样本种类丰富、样本彼此的相似度很高,所以各深度模型在该数据集上的表现都很好.但从表8可以看出,在交叉验证下,本文所提模型在该数据集上的表现更为稳定一些.
与当前前沿模型相比,本文所提模型在4个公开数据集上均能取得最佳的识别效果,一定程度上可以验证其优越性.为了更好地展示模型的实际价值,本文将在3.4节根据具体的医学案例,介绍所提模型的使用方法并给出仿真实验的结果.
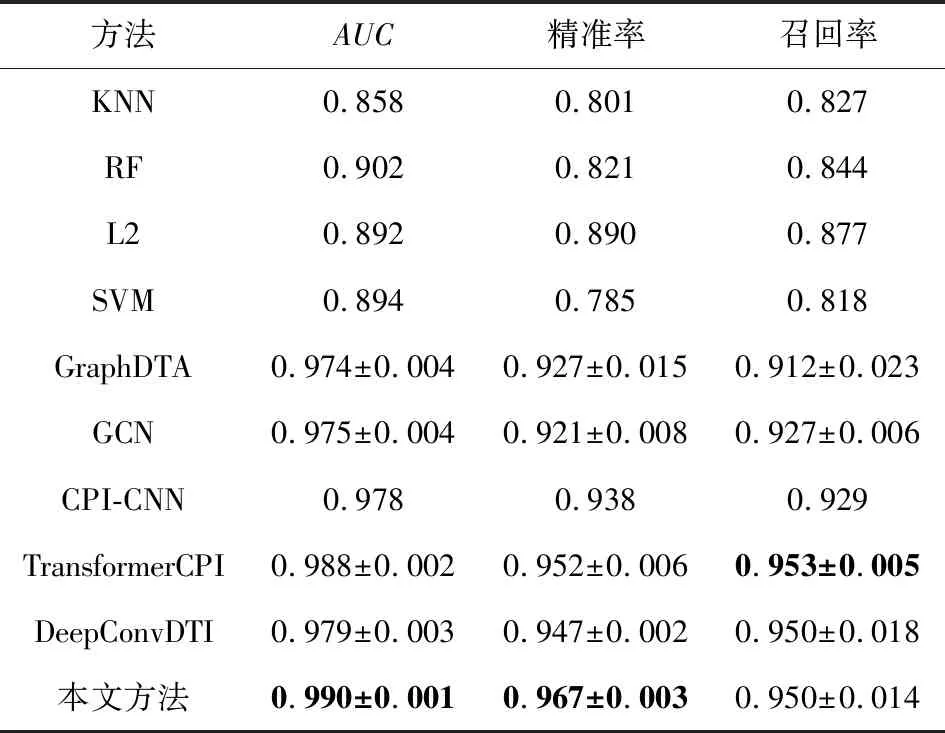
Table 8 Comparative Experiments on C.elegans Dataset表8 C.elegans数据集上的对比实验
3.4 模型在筛选治愈阿尔兹海默症药物的应用
痴呆症是公共卫生管理中最值得关注的问题之一,而80%以上的痴呆症患者患有阿尔茨海默病(AD)[44].然而,当前普及的治疗方案仅可起缓解作用,无法根据病理学使患者痊愈或逆转患者的疾病进程.因此,当前医学界仍急需一种可延缓或阻止疾病进展的新型治疗方案.神经递质乙酰胆碱(ACh)的减少导致胆碱能神经元的丢失在医学界是公认的AD病因,而通过抑制乙酰胆碱酯酶(AChE)可以提升ACh的水平,以此提高患者的认知能力[43].
虽然乙酰胆碱酯酶(AChE)在疾病晚期的活性会降低,但与图7所示的AChE结构类似的丁酰胆碱酯酶(BuChE)含量在疾病晚期会有增加的可能[36],而BuChE的含量增加也会水解ACh从而再次加重患者的病情.敲除乙酰胆碱酯酶基因的小鼠实验[45]支持了这一假设,也证实了通过抑制BuChE可以改善认知能力以及提升患者的记忆力.因此,寻找抑制AChE和BuChE的靶向药物是治疗AD的关键方向.根据这一理论,本文将所提出的模型设计成一套可应用的药物筛选工具[46],并用之筛选抑制AChE和BuChE的药物.具体操作步骤:1)将待测蛋白质序列数据与大量的药物分子式数据同时放入筛选工具中;2)系统会根据训练好的模型对药物进行筛选;3)系统会给出Top15的药物序号,并将这些药物与待测蛋白质相互作用的预测结果以直方图的形式自高而低展现.为了证实文本所提模型的价值,在该项任务中选择用于测试的药物分子均不在训练集中,但需要指明的是,训练集中包含AChE的序列数据但不包含BuChE的序列数据,而AChE序列与BuChE序列相似性为65%.当然,PDI预测模型的核心原理是通过学习已有的蛋白质药物相互作用关系并从中拟合出一个规律函数来推断未知的相互作用关系.所以,如果测试的样本与训练的内容毫无关联,那么其推断的依据便失去了理论支撑.
Fig. 7 Mimic diagram of protein-drug interaction图7 蛋白质-药物相互作用模拟图
本文借用Kumar等人[47]提供的药物数据作为测试的目标,测试数据中包含的35种化合物是由Kumar等人[47]在Asinex库中手动筛选出的,具有高度可信性.文献[47]中给出了药物分子的2维结构式以及Asinex编号,本文通过相关编号以及药物的结构式在PubChem上检索出用于预测PDI的SMILES式.同时,Kumar给出了这部分药物分子对AChE和BuChE的抑制率(inhibition rate,IR),本文按照其定义的准则,将IR<0.5记为无相互作用,IR>0.5记为有相互作用,获得的最终测试数据如表9所示.
值得注意的是,测试药物皆不存在于训练集中,但训练集中不排除具有相似结构以及相同官能团的药物分子.根据系统的预测结果,本文按预测值高低将分子序号排列,得到Top15的直方图.如图8所示,其中灰色直方图表示在原数据中该药物与测试酶具有相互作用,条纹直方图表示原数据中该药物与测试酶没有相互作用.
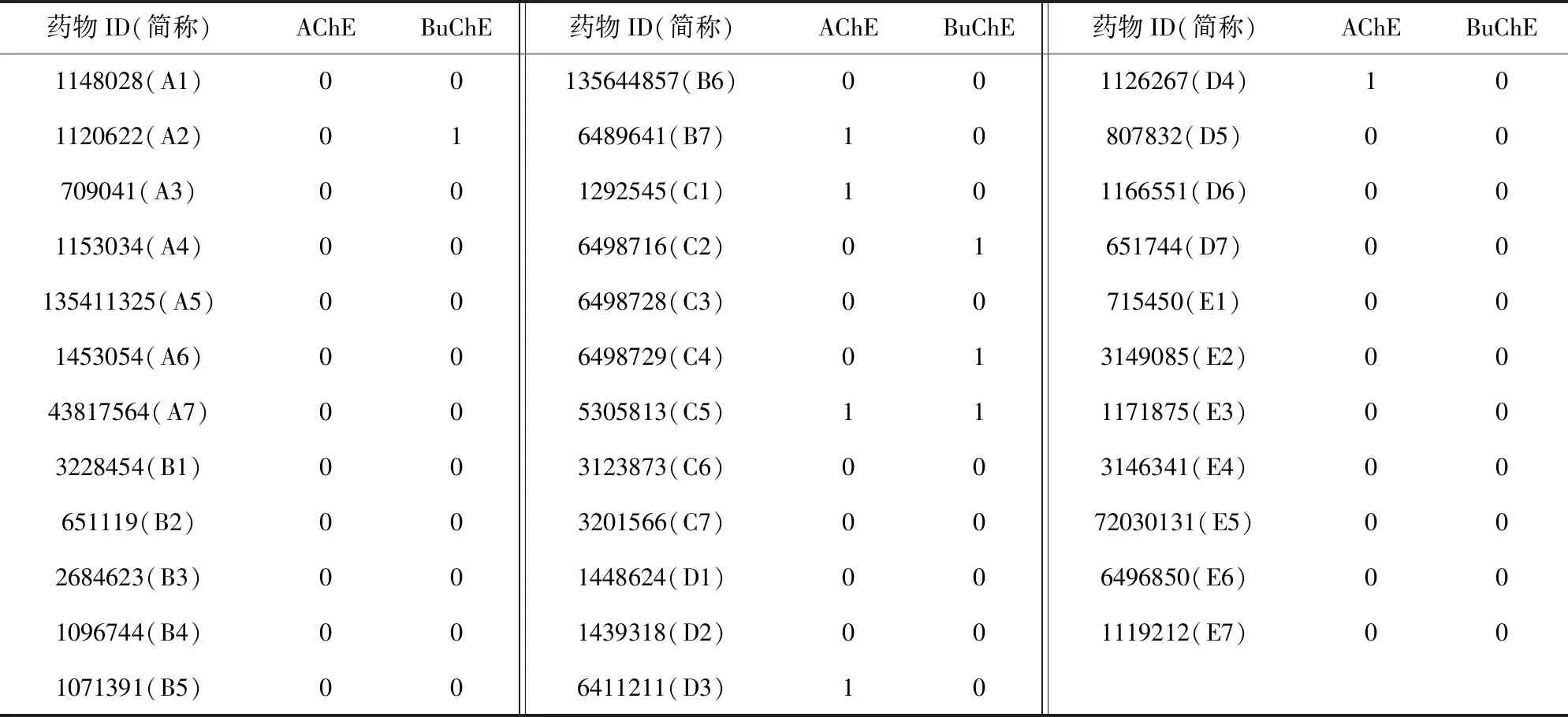
Table 9 The Manual Screening Drug Data Sets of AChE and BuChE Inhibition表9 手动筛选的AChE和BuChE抑制药物数据集
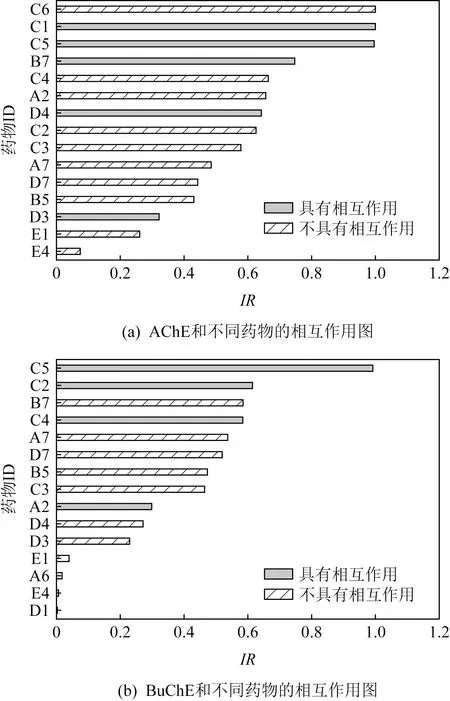
Fig. 8 The test results of protein-drug interaction图8 蛋白药物相互作用测试结果
由图8可以看出,有相互作用的蛋白质药物组合基本存在于预测的Top15范围内,说明本文提供的模型具有实际的应用性.但不排除部分仅可抑制丁酰胆碱酯酶的药物在预测和乙酰胆碱酯酶相互作用时成正相互作用,这在所难免.具备相似氨基酸序列的蛋白质,其与药物相互作用的预测值也十分贴近,近似结构蛋白具备近似生物活性是预测PDI的重要理论支撑点但也是难以区分细节差异的原因,同时也是当前深度模型仅能做药物筛选而不能做药物设计的原因.想实现端到端模型设计药物分子还需更深入的研究.
4 结 论
本文提出了一种基于自注意力机制和多药物特征融合的蛋白质-药物相互作用预测算法.首先,合理融合基于药物分子结构特征的Morgan指纹、Mol2Vec表示向量以及消息传递网络所提特征;随后,将融合结果对由密集型卷积所提取的蛋白特征做注意力加权;最后综合两者特征,利用自注意力机制和双向门控循环单元预测蛋白质药物相互作用.本文在BindingDB,Kinase,Human,C.elegans这4个数据集上进行验证,无论是与传统机器学习算法相比还是与当前各深度学习算法相比,本文的算法都明显更优.同时,本文根据所训练模型针对抑制丁酰胆碱酯酶和乙酰胆碱酯酶做药物筛选实验.结果表明,本文所提模型具备可靠的应用性,但依然存在提升空间.首先,与其他基于深度学习的方法类似,黑盒模型依旧缺乏可解释性[48].其次,利用同源蛋白以及相似结构药物具备相似生物活性原理预测PDI和解决由细节差异带来药性影响的问题存有矛盾,当前还缺乏对此的深入研究.未来,会基于这2个问题继续探索,不断为药物筛选工作提供新的解决方案.
作者贡献声明:华阳负责数据采集、实验设计、编程实现和论文撰写;李金星负责数据采集和实验设计;冯振华参与论文修改;宋晓宁负责实验指导,参与论文修改;孙俊参与论文修改;於东军参与实验指导和论文修改.
猜你喜欢
今日农业(2022年15期)2022-09-20
农业工程学报(2022年12期)2022-09-09
计算技术与自动化(2022年1期)2022-04-15
海外星云(2021年9期)2021-10-14
上海师范大学学报·自然科学版(2019年5期)2019-12-13
中国新通信(2017年9期)2017-05-27
新课程学习·中(2013年3期)2013-06-14
中学数学研究(2008年3期)2008-12-09
