
荧光吡唑酰胺二唑类衍生物的合成、表征及抗菌活性研究
2022-09-02 11:28陈连清雷金澌王川川吴忠达杜艳婷
中南民族大学学报(自然科学版) 2022年5期
陈连清,雷金澌,王川川,吴忠达,杜艳婷
(中南民族大学 化学与材料科学学院& 催化转化与能源材料化学教育部重点实验室,武汉430074)
在现代农药的发展中,吡唑类化合物表现了出色的杀虫性能[1],被用于消灭不利于农作物生长的害虫如蚜虫、赤松毛虫、小菜蛾[2]等,也具有出色的抗菌性能,尤其是对普通变形杆菌、金黄色葡萄球菌和大肠埃希菌具有出色的杀菌效果[3].除此之外,该化合物在抗病毒[4]、抗炎、抗溃疡及抗癌[5-6]等方面也有着良好的活性,还具有优异的热稳定和抗氧化能力[7],在新药设计中常被用作药效基团,具有低毒、不挥发、不易燃和可生物降解等优点,还可作为反应介质和相转移.酰胺类化合物在农药的应用中也发挥着优秀的作用,利用了酰胺类化合物的水溶性[8]、生物活性[9]和选择性[10]的一系列优点,与吡唑类化合物相结合,合成了吡唑酰胺类化合物,由于其容易形成配位键,表现出多种配位的性能,也能够形成氢键,这类衍生物被应用于灭菌、抗病毒、促进植物生长[11]、除虫、除草[12]等多个方面.二唑类衍生物具有荧光性[13]和良好的光敏性质[14],能够发出蓝色荧光,可用于生产荧光剂、闪烁剂,并且作为高分子感光材料,能制成发蓝光的有机光电薄膜,被广泛应用于电致发光仪器中,其拥有强的电子亲和势可在电致发光器件中被用作电子传输材料.而其作为杀虫农药时,可使用荧光灯以检测出是否有残留,具有荧光性的物质能够在光照作用下进行自降解[15],有利于后期对农药的处理,不会造成污染,对环境保护起到很好的效果,因此具有荧光性的物质作为农药对虫害进行综合治理具有很大的应用前景,被广泛应用于农药领域.
雷英杰等[16]对噁二唑基团与硫醚酰胺进行拼接,测试其抗菌性能,表现出很好的杀菌效果,但合成噁二唑酰胺类化合物是以PEG-400为相转移催化剂,乙腈为溶剂,利用超声波辐射相转移方法合成,这一方法的缺点是能耗较高.苏诗淼等[17]通过Strecker 反应,经无水Zn 或I2的催化下,将α-氨基腈类活性基团引入吡唑胺底物中,获得含氰基的多取代吡唑衍生物,但合成该类衍生物时生成的中间体需要在高温下真空活化,操作复杂而后处理麻烦,且产率有待提高.这类研究中大多化合物仅仅将一类基团进行衍生,或将两类基团进行简单拼接,且化合物结构较为固定,产物抗菌的菌种种类数较少,化学性质或物理用途较为单一,且这些合成方法大都所需能耗大、时间长、产率有待提高、后处理麻烦、溶剂易挥发,在一定程度上限制了对相关类型化合物的应用.
基于上述缺点,本文对实验进行了优化改进,使用一种新的合成方法即微波水热合成法,将上述三类基团成功连接在一起.微波水热法利用高频震荡的微波能量场下使分子运动,运动过程中将动能转化为热能,可以在分子水平上搅拌以实现对反应物加热的效果,即无梯度加热,克服了常规水热容器加热不均匀的特点,微波水热合成法对有机物的合成具有出色的效果.结果证明:该法具有产率高、时间短、操作简便快捷、节约能源、保护环境等优点,在作为绿色农药时能够有效自降解,可大量制备,具有十分非常广阔的应用前景.
为了找到具有荧光性的绿色农药,在吡唑酰胺类化合物中引入了二唑(噁二唑和噻二唑)基团.利用胺、2,3-二氰基-丙酸乙酯、醇、酰卤等一系列基本原料,合成氰基吡唑,然后经酯化、酰肼化、酰胺化后,再利用微波水热合成法与环合剂反应,合成具有荧光性和抗菌活性的吡唑酰胺二唑类衍生物,其结构式如图1所示.

图1 吡唑酰胺二唑类衍生物的结构通式Fig.1 General structure of pyrazole amide diazole derivatives
图1 中,M 为—O—时,命名为N-(5-[1,3,4]噁二唑-2-基-2H-吡唑-3-基)-酰胺类衍生物,M为—S—时,命名为N-(5-[1,3,4]噻二唑-2-基-2H-吡唑-3-基)-酰胺类衍生物;R1、R2为烷基、卤代烷基、苯基、苄基等的一种.化学结构方面,可合成符合该通式的一大类衍生物,活性基团种类较多,并且能对更多种类的菌株起到抑制效果,这是一类综合性的在光电领域及医药、农药、抗菌消炎杀毒等领域具有多功能途的化学物质,应用领域更为广阔.
1 实验部分
1.1 材料和仪器
硝酸钠、浓硫酸、环己胺、苯胺、对羟基苯胺、对硝基苯胺、对氯苯胺、对甲氧基苯胺、对氟苯胺、5-氨基吲哚、乙酸、甲苯、硫酸镁、乙醇、三氯化铁、乙酸乙酯、四氢呋喃、石油醚、水合肼、乙酰氯、丙酰氯、丁酰氯、苯甲酰氯、苯甲酰溴、对羟基苯甲酰溴、吡啶、三氯氧磷、DMSO、氨水、五氧化二磷、五氧化二钒,试剂和药品均为市售化学纯度,使用前未经进一步纯化.
微波水热仪(MDS-10 型,上海新仪微波化学);核磁共振谱仪(AVANCE III 400M 型,德国Bruker);元素分析仪(Vario-EL III CHNS 型,北京来亨科贸);傅立叶红外光谱仪(NEXUS-470 智能型,美国Nicolet);荧 光 光 谱 仪(PE LS-55 型,美 国PerkinElmer);紫外-可见光谱分光光度计(Perkin-Elmer Lambda-Bio35,日本岛津);熔点仪(MPA100,美国Optimelt);电子天平(CP114,上海奥豪斯仪器).
1.2 吡唑酰胺二唑类衍生物的合成
吡唑酰胺二唑类衍生物的合成方法流程如图2所示.

图2 吡唑酰胺二唑类衍生物的合成路线图Fig.2 Synthetic route of pyrazole amide diazole derivatives
N-(5-[1,3,4]噁二唑-2-基-2H-吡唑-3-基)-酰胺类衍生物的合成如下.
步骤1:将9.8 g NaNO2加入到三口烧瓶中,在冰浴条件下,加入35.0 mL 浓硫酸和35.0 mL 冰乙酸混合溶液,分别滴加环己胺、苯胺、对羟基苯胺和对硝基苯胺(30.0 g)的冰乙酸溶液.缓慢升温,滴加2,3-二氰基丙酸乙酯(20.0 g)的冰乙酸溶液,持续搅拌5 h.甲苯萃取,向有机层中加入20 mL浓氨水,磁力搅拌2 h.静置分层后用无水MgSO4干燥,旋转蒸发,利用甲苯将粗产物中重结晶,析出得到ⅠO1~ⅠO5晶体,这几步的产率范围:86.2%~92.0%.ⅠO1 为5-氨基-1-环己基-1H-吡唑-3-甲腈,ⅠO2 为5-氨基-1-苯基-1H-吡唑-3-甲腈,ⅠO3 为5-氨基-1-苯基-1H-吡唑-3-甲腈,ⅠO4 和ⅠO5 为5-氨基-1-(4-硝基-苯基)-1H-吡唑-3-甲腈.将原料改用对氯苯胺、对甲氧基苯胺、对氟苯胺和5-氨基吲哚制备得到ⅠT1~ⅠT5,这几步的产率范围:87.1%~92.6%.ⅠT1为5-氨基-1-(4-氯-苯基)-1H-吡唑-3-甲腈,ⅠT2和ⅠT3为5-氨基-1-(4-甲氧基-苯基)-1H-吡唑-3-甲腈,ⅠT4 为5-氨基-1-(4-氟-苯基)-1H-吡唑-3-甲腈,ⅠT5 为5-氨基-1-(1H-吲哚-4-基)-1H-吡唑-3-甲腈.其结构经熔点、傅里叶红外、核磁共振氢谱方法检测确证.
步骤2:在三颈烧瓶中将15 mmol ⅠO1~ⅠO5,30 mL无水乙醇和15 mmol三氯化铁混合,放入搅拌磁子,在90 ℃下搅拌回流反应12 h,用水和乙酸乙酯萃取,旋蒸后,用体积比1∶4的乙酸乙酯和石油醚用作洗脱液进行硅胶柱层析分离、旋蒸、干燥得到氰基酯化产物.
步骤3:在三颈烧瓶中将20 mmol上述氰基酯化产物与30 mL的无水乙醇混合,放入搅拌磁子,磁力搅拌10 min 后缓慢滴加60 mmol 80%的水合肼溶液,并在80 ℃下加热回流3 h,待反应成功完成后,旋蒸,过滤得到滤饼,少量无水乙醇洗涤滤饼并干燥,得到白色固体酰肼化合物.
步骤4:于通风橱中取0.01 mol 上述酰肼化合物,用30 mL四氢呋喃溶解,取将0.02 mol NaH,进行少量多加入其中.磁力搅拌10 min,然后分别加入10 mmol 乙酰氯、丙酰氯、丁酰氯、乙酰氯、苯甲酰氯和对羟基苯甲酰溴,85 ℃下加热回流14 h,用乙酸乙酯和水萃取后,干燥,用体积比1∶4的乙酸乙酯和石油醚用作洗脱液进行硅胶柱层析分离,然后旋转蒸发、干燥,制备化合物ⅡO 和ⅡT,其结构经熔点、傅里叶红外、核磁共振氢谱方法检测确证.
步骤5:分别将25 mL 吡啶、15 mmol 化合物ⅡO和15 mmol 环合剂五氧化二磷加入100 mL 反应釜中,将反应釜密封置于微波反应器内,设置温度为230 ℃,并使用500 W 功率进行微波水热辐射,持续6 h,待其自然冷却至室温,将所得混合物用蒸馏水和无水乙醇洗涤多次,干燥,然后用无水乙醇重结晶,得到最终产物噁二唑类衍生物ⅢO1~ⅢO5,化合物ⅡT 中加入的环合剂一律为五硫化二磷,控制其他反应条件与方法不变,得到最终产物噻二唑类衍生物ⅢT1~ⅢT5.所有最终产物相关物性常数及表征数据如下.
化合物N-[5-(5-甲基-[1,3,4]噁二唑-2-基)-2-环己基-2H-吡唑-3-基]-乙酰胺(ⅢO1),产率97.9%,m.p.178.4~179.3 ℃;1H NMR(CDCl3,400 MHz)δ:8.29(s,1H,N-H),6.16(s,2H,C-H),3.76(m,1H,C-H),2.37(s,3H,C-H3),2.08(s,2H,C-H3),1.75(m,4H,C-H2),1.39(m,4H,CH2),1.23(m,1H,C-H2).Anal.calcd for C14H19N5O2:C 58.13,H 6.57,N 24.22;found C 58.22,H 6.73,N 24.19.
化合物N-[5-(5-乙基-[1,3,4]噁二唑-2-基)-2-苯基-2H-吡唑-3-基]-丙酰胺(ⅢO2),产率96.6%,m.p.188.5~189.3 ℃;1H NMR(CDCl3,400 MHz)δ:8.20(s,1H,N-H),7.58(d,J=7.2 Hz,2H,Ar-H),7.43(t,J= 7.6 Hz,2H,Ar-H),7.32(d,J=7.6 Hz,1H,Ar-H),6.66(s,1H,C-H),2.67(m,2H,C-H2),2.24(m,2H,C-H2),1.32(t,J= 8.0 Hz,3H,C-H3),1.23(t,J= 8.0 Hz,3H,C-H3).Anal.calcd for C16H17N5O2:C 61.74,H 5.47,N 22.51;found C 61.68,H 5.39,N 22.44.
化合物N-[2-(4-羟基-苯基-5-(5-丙基-[1,3,4]噁二唑-2-基)-2H-吡唑-3-基]-丁酰胺(ⅢO3),产率96.6%,m.p.195.3~196.1 ℃;1H NMR(CDCl3,400 MHz)δ:8.16(s,1H,N-H),7.36(t,J= 8.0 Hz,2H,Ar-H),6.94(d,J= 7.6 Hz,2H,Ar-H),6.71(s,1H,C-H),5.23(s,1H,C-H),2.69(t,J= 7.2 Hz,2H,C-H2),2.31(t,J= 7.2 Hz,2H,C-H2),1.80(m,2H,C-H2),1.69(m,2H,C-H2),0.97(t,J= 8.0 Hz,3H,C-H3),0.92(t,J= 8.0 Hz,3H,C-H3).Anal.calcd for C18H21N5O3:C 60.85,H 5.92,N 19.72;found C 60.77,H 5.99,N 19.59.
化合物N-[5-(5-甲基-[1,3,4]噁二唑-2-基)-2-(4-硝基-苯基)-2H-吡唑-3-基]-乙酰胺(ⅢO4),产率96.8%,m.p.216.2~216.9 ℃;1H NMR(CDCl3,400 MHz)δ:8.41(d,J= 8.4 Hz,2H,Ar-H),8.12(s,1H,N-H),7.75(d,J= 7.6 Hz,2H,Ar-H),6.92(s,H,C-H),2.59(s,3H,C-H3),2.16(s,3H,CH3).Anal.calcd for C14H12N6O4:C 51.22,H 3.66,N 25.61;found C 51.34,H 3.59,N 25.49.
化合物N-[2-(4-硝基-苯基)-5-(5-苯基-[1,3,4]噁二唑-2-基)-2H-吡唑-3-基]-苯甲酰胺(ⅢO5),产 率96.7%,m.p.217.6~218.5 ℃;1H NMR(CDCl3,400 MHz)δ:8.15(s,1H,N-H),7.95(t,J= 7.2 Hz,2H,Ar-H),7.96(d,J= 7.2 Hz,2H,Ar-H),7.71(d,J= 7.6 Hz,2H,Ar-H),7.55(m,2H,Ar-H),7.54(t,J= 7.2 Hz,2H,Ar-H),7.52(t,J= 8.0 Hz,2H,Ar-H),7.49(t,J= 8.0 Hz,2H,Ar-H),6.76(s,1H,C-H).Anal.calcd for C24H16N6O3:C 66.06,H 3.67,N 19.27;found C 66.31,H 3.80,N 19.44.
化合物N-[2-(4-氯-苯基)-5-(5-苯基-[1,3,4]噻二唑-2-基)-2H-吡唑-3-基]-苯甲酰胺(ⅢT1),产率95.0%,m.p.225.3~226.0 ℃;1H NMR(CDCl3,400 MHz)δ:8.27(s,1H,N-H),7.96(t,J= 7.6 Hz,2H,Ar-H),7.52(m,1H,Ar-H),7.55(d,J=7.6 Hz,2H,Ar-H),7.41(m,2H,Ar-H),7.39(m,2H,Ar-H),7.37(d,J= 8.4 Hz,2H,Ar-H),7.22(m,1H,Ar-H),7.27(d,J= 8.0 Hz,2H,Ar-H),6.79(s,1H,C-H).Anal.calcd for C24H16ClN5OS:C 63.65,H 3.54,N 15.47;found C 63.37,H 3.41,N 15.09.
化合物N-[2-(4-甲氧基-苯基)-5-(5-苯基-[1,3,4]噻二唑-2-基)-2H-吡唑-3-基]-苯甲酰胺(ⅢT2),产率94.7%,m.p.235.7~236.4 ℃;1H NMR(CDCl3,400 MHz)δ:8.23(s,1H,N-H),7.96(t,J= 8.0 Hz,2H,Ar-H),7.51(m,1H,Ar-H),7.54(d,J= 8.0 Hz,2H,Ar-H),7.41(m,2H,Ar-H),7.32(m,2H,Ar-H),7.27(m,2H,Ar-H),7.22(d,J= 8.4 Hz,1H,Ar-H),6.93(d,J= 8.8 Hz,2H,Ar-H),6.71(s,1H,C-H),3.83(s,3H,CH3).Anal.calcd for C25H19N5O2S:C 66.23,H 4.19,N 15.45;found C 66.28,H 4.29,N 15.39.
化合物N-[5-[5-(4-羟基-苯基)-[1,3,4]噻二唑-2-基]-2-(4-甲氧基-苯基)-2H-吡唑-3-基]-苯甲酰胺(ⅢT3),产率93.9%,m.p.238.5~239.4 ℃;1H NMR(CDCl3,400 MHz)δ:8.28(s,1H,N-H),7.98(t,J= 7.2 Hz,2H,Ar-H),7.55(m,1H,Ar-H),7.44(m,2H,Ar-H),7.39(d,J=7.6 Hz,2H,Ar-H),7.23(d,J= 7.6 Hz,2H,Ar-H),6.96(d,J= 8.4 Hz,2H,Ar-H),6.75(d,J= 8.0 Hz,2H,Ar-H),6.71(s,1H,C-H),5.23(s,1H,C-H),3.83(s,3H,C-H3).Anal.calcd for C25H19N5O3S:C 63.97,H 4.05,N 14.93;found C 63.65,H 4.09,N 14.89.
化合物N-[2-(4-氟-苯基)-5-[5-(4-羟基-苯基)-[1,3,4]噻二唑-2-基]-2H-吡唑-3-基]-苯甲酰胺(ⅢT4),产率94.7%,m.p.247.6~248.3 ℃;1H NMR(CDCl3,400 MHz)δ:8.27(s,1H,N-H),7.98(t,J= 8.0 Hz,2H,Ar-H),7.56(m,1H,Ar-H),7.43(m,2H,Ar-H),7.37(d,J= 7.2 Hz,2H,Ar-H),7.21(d,J= 8.0 Hz,2H,Ar-H),7.12(d,J= 8.0 Hz,2H,Ar-H),6.86(d,J= 8.4 Hz,2H,Ar-H),6.74(s,1H,C-H),5.26(s,1H,O-H).Anal.calcd for C24H16FN5O2S :C 63.02,H 3.50,N 15.32;found C 63.21,H 3.68,N 15.41.
化合物N-[5-[5-(4-羟基-苯基)-[1,3,4]噻二唑-2-基]-2-(1H-吲哚-4-基)-2H-吡唑-3-基]-苯甲酰胺(ⅢT5)产率95.8%,m.p.256.8~257.4 ℃;1H NMR(CDCl3,400 MHz)δ:10.21(d,J= 8.8 Hz,1H,N-H),8.24(s,1H,N-H),7.98(t,J=7.2 Hz,2H,Ar-H),7.54(m,1H,Ar-H),7.48(m,2H,Ar-H),7.43(d,J= 7.6 Hz,2H,Ar-H),7.39(d,J=7.6 Hz,1H,N-H),7.35(d,J=7.6 Hz,1H,Ar-H),7.27(m,1H,Ar-H),7.13(d,J= 8.0 Hz,1H,Ar-H),6.85(d,J= 8.0 Hz,2H,Ar-H),6.77(s,1H,C-H),6.47(d,J= 7.6 Hz,1H,Ar-H),5.21(s,1H,O-H).Anal.calcd for C26H18N6O2S:C 65.27,H 3.77,N 17.57;found C 65.42,H 3.77,N 17.34.
1.3 发光性能测试
为了测试吡唑酰胺二唑类衍生物的发光性能,测试N-(5-[1,3,4]噁二唑-2-基-2H-吡唑-3-基)-酰胺类衍生物(ⅢO)、N-(5-[1,3,4]噻二唑-2-基-2H-吡唑-3-基)-酰胺类衍生物(ⅢT)的荧光、紫外-可见光谱,对光物理性质进行分析.利用二氯甲烷配制浓度为50 mg·L-1的化合物ⅢO1~ⅢO5、ⅢT1~ⅢT5 溶液各250 mL 作为供试样品,使用荧光光谱仪射测试其荧光发射波长和荧光量子产率;紫外-可见光谱分光光度计测定紫外-可见吸收.
1.4 抗菌活性测试
为了测试吡唑酰胺二唑类衍生物的抗菌活性,对抗金黄色葡萄球菌活性进行评价.
分别将供试化合物ⅢO1~ⅢO5、ⅢT1~ⅢT5溶于二甲基亚砜,预配成0.1%的浓度,然后用1%乙酸蒸馏水溶液稀释至4 种浓度,分别为100、10、1、0.1 mg·L-1,将其用作抗菌剂,并与阳性对照药诺氟沙星(NF)的抗菌效果作比较.直接用1%乙酸蒸馏水溶液配制成10、1、0.1 mg·L-13 种浓度,制备一组1%的乙酸蒸馏水溶液作为空白对照.
使用平皿测试法进行对金黄色葡萄球菌的抗菌活性进行研究,准备有金黄色葡萄球菌的牛肉膏蛋白的胨培养基,将直径为5 cm 的消菌滤纸片分别浸入上述步骤配制的不同化合物的不同浓度抗菌剂中,将这些纸片贴到培养基上,转移至恒温培养基中,设置温度为37 ℃,培养24 h 后取出圆形滤纸片并测量抑菌圈直径.与对照药物诺氟沙星相比较,对该类衍生物的抗菌活性进行评估.
2 结果与讨论
2.1 产物的结构表征
原料通过酯化、酰肼化、酰胺化后得到化合物Ⅱ,再利用微波水热合成法与环合剂反应进行调控合成10 种含二唑单元的N-(5-[1,3,4]二唑-2-基-2H-吡唑-3-基)-酰胺类衍生物,取得了理想效果.产物产率由取代基团的诱导效应和共轭效应相互影响,随着引入具有强推电子的基团(—OH、—OCH3)时,产率略有降低,当取代基的位阻效应和共轭程度大时,产率有所下降.依据10 类产物的核磁氢谱数据可知:芳环δH为8.1~8.4;δH在3.83 时,可归属为—OCH3的H 的特征化学位移;δH在5.2 时,可归属为—OH 中H 的特征化学位移.各种化合物相应的核磁氢谱和元素分析数据,表明所得结果与设计的目标化产物的结构相一致.
2.2 不同合成条件及方法的影响
2.2.1 反应时间对微波水热法得到产率的影响
控制步骤5 的反应温度为230 ℃,测试反应时间分别为0.5、1、2、3、4、5、6、7、8 h 时对微波水热法制备N-[5-(5-乙基-[1,3,4]噁二唑-2-基)-2-苯基-2H-吡唑-3-基]-丙酰胺(ⅢO2)的影响,表1为不同反应时间下得到的ⅢO2 的产率.由表1 可见:反应时间对反应产率有影响,随着反应时间的增加,ⅢO2的产率先逐渐增加然后保持在较高范围,反应时间在3~6 h 之间产率较高并且变化范围很小,反应时间过长时产率出现下降的现象,微波水热法进行实验的最佳的反应时间为6 h,达到了97.9%的产率.

表1 不同反应时间下ⅢO2的产率Tab.1 ⅢO2 production rate under different reaction time
2.2.2 反应温度对不同合成方法制备得到产率的影响
与上述处理方法相同,不同的是步骤5 中将混合物分别利微波水热法、用水热法、烘箱加热法、微波辐射法和油浴加热的方法合成,并将反应温度分别设置在80、110、140、170、200、230、260 ℃.
水热法:将15 mmol 化合物ⅡO2、25 mL 蒸馏水和15 mmol 五氧化二磷混合均匀,加入到在100 mL 高压反应釜中,密封置于烘箱中,调节温度为上述反应温度,调节反应时间为6 h,待反应结束后关闭反应器,冷却后用蒸馏水洗涤混合物,干燥,无水乙醇重结晶,称重,计算产物收率.
烘箱加热法:在手套箱中称取15 mmol 化合物ⅡO2 、25 mL 吡啶和15 mmol 五氧化二磷,用玛瑙研钵混合均匀,研磨至呈粉末状,加入到250 mL 的高压反应釜中,拧紧密封盖并置入烘箱中,调节温度为上述反应温度,反应时间为6 h,待反应结束后关闭反应器,自然冷却到室温,用蒸馏水洗涤混合物,干燥,无水乙醇重结晶,称重,计算产物收率.
微波辐射法:在100 mL 三颈烧瓶中加入15 mmol 化合物ⅡO2、25 mL 吡啶和15 mmol 五氧化二磷,然后将三颈烧瓶放入微波反应器中,500 W 功率下辐射6 h,待自然冷却至室温,用蒸馏水洗涤混合物,干燥,无水乙醇重结晶,称重,计算产物收率.
油浴加热法:在100 mL 三颈烧瓶中加入15 mmol 化合物ⅡO2、25 mL 吡啶和15 mmol 五氧化二磷,油浴加热回流6 h,冷却后用蒸馏水洗涤混合物,干燥,无水乙醇重结晶,称重,计算产物收率.
表2 为不同合成方法在不同反应温度下得到的ⅢO2 的产率.由表2 可见:反应温度对不同合成方法制备得到的产率都有着不同程度的影响,升高反应温度,产率均随着增加,在温度为230 ~260 ℃时产率保持在一定范围,该反应温度为合成该类化合物的最佳反应温度.表2 中发现微波水热法能得到最高的产率,达到了97.0%,相比于另外四类合成方法,微波水热法具有较广的应用前景.

表2 不同合成方法在不同反应温度下ⅢO2的产率Tab.2 Yield of ⅢO2 in different synthetic methods at different reaction temperatures
2.2.3 反应时间对不同合成方法制备得到产率的影响
控制反应温度150 ℃,反应时间分别为0.5、1、2、4、6、8、12 h 时对不同合成方法制备ⅢO2 的影响,表3 为不同反应时间下得到的ⅢO2 的产率.由表3 可见:反应温度对不同合成方法制备得到的产率都有着不同程度的影响,其中微波水热法的反应最迅速,随着反应时间增加,产率不断提高,在反应时间到6 h 时反应速度下降,反应到12 h 的产率相比于6 h 区别不大,仅增加了1.7%.为了节约能源,将其作为该条件下的最佳反应时间,达到了90.1%的产率,其他方法的产率也与反应时间有关,但反应12 h 后得到的产率明显低于微波水热法.利用微波水热法能够促进反应进行,节省反应时间,还有效地提升了反应速率与产率.

表3 不同合成方法在不同反应时间下ⅢO2的产率Tab.3 Yield of ⅢO2 in different synthetic methods at different reaction times
2.3 发光性能评价
化合物ⅢO1~ⅢO5、ⅢT1~ⅢT5 溶液的荧光发射和紫外-可见吸收数据如表4 所示.由表4可见:当改变取代基R1和R2种类,化合物不同类型的取代基,具有推动和拉动电子的不同感应效应,将会得到不同的荧光发射和紫外-可见吸收波长,不同的电子效应与共轭程度也对其有影响,当具有更大共轭体系时,荧光发射和紫外-可见吸收波长会进一步红移.在具有大π 键结构的共轭体系化合物中,紫外-可见吸收与荧光发射随离域电子的愈易被激发而愈易产生.由荧光测试结果可见:这些吡唑酰胺二唑类衍生物都具有较高的荧光量子产率,所有样品的荧光产率均在40%以上,能有效地将吸收的光能转变成荧光.总结得出:10 个制备的样品均有紫外-可见吸收性能和良好的荧光发射能力,表现出优秀的光物理性能,应用前景十分广阔.

表4 化合物ⅢO1~ⅢO5和ⅢT1~ⅢT5的紫外-可见吸收和荧光发射数据Tab.4 UV-visible absorption data and fluorescence emission of compounds ⅢO1~ⅢO5 and ⅢT1~ⅢT5
2.4 抗菌活性评价
化合物ⅢO1~ⅢO5 和ⅢT1~ⅢT5 对金黄色葡萄球菌的抗菌效果如表5 所示.由表5 可见:与诺氟沙星(NF)的抗菌效果相比,化合物ⅢO1~ⅢO5 和ⅢT1~ ⅢT5 的抗金黄色葡萄球菌(Staphylococcus aureusCMCC26003)活性均表示较高活性或中等活性,随着被稀释程度增加,所有化合物的抗菌活性减弱,在达到0.1 mg·L-1时,抗金黄色葡萄球菌活性中等,但仍有可观的活性,可见N-(5-[1,3,4]二唑-2-基-2H-吡唑-3-基)-酰胺类衍生物具有较好的抗菌活性.
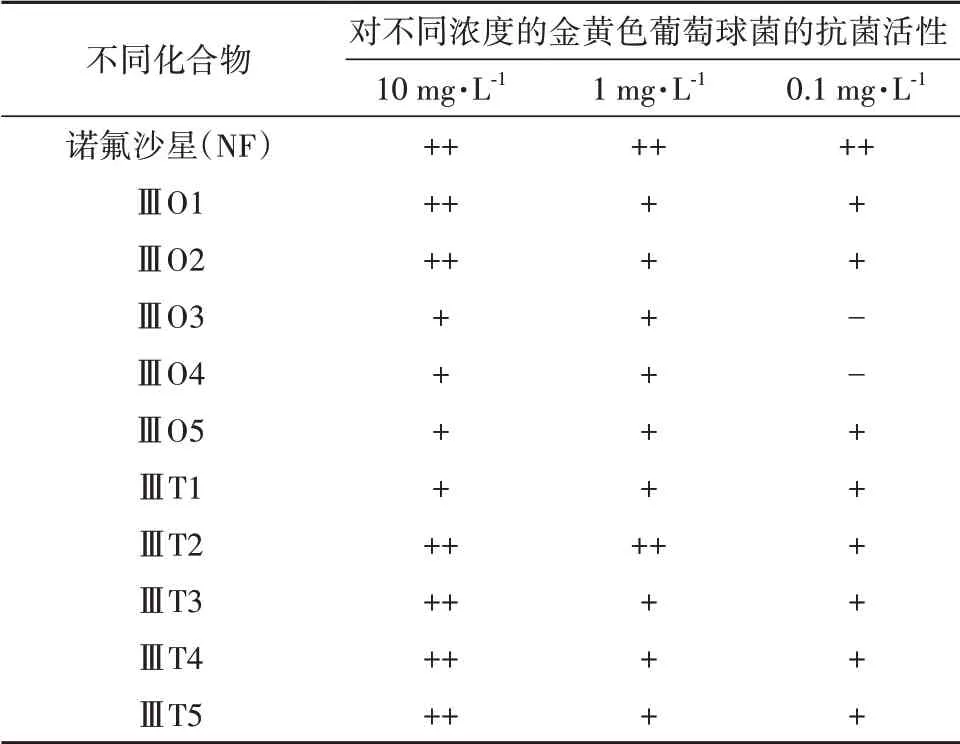
表5 化合物ⅢO1~ⅢO5和ⅢT1~ⅢT5对金黄色葡萄球菌的抗菌活性Tab.5 Antibacterial activity of compounds ⅢO1~ⅢO5 andⅢT1~ⅢT5 against Staphylococcus aureus
3 结语
利用改进的帕尔-克诺尔(Paal-Knorr)反应,以一系列胺类化合物为初始原料经酯化、酰肼化、酰胺化,再利用微波水热合成法与环合剂反应,合成了一系列具有荧光性和抗菌活性的吡唑酰胺二唑类衍生物.与其他制备方法相比,微波水热合成法是一种易于高效制备、节能环保、生产成本相对较低的合成方法,有望成为高效制备吡唑酰胺二唑类衍生物的新方法,应用前景十分广阔.化学结构方面,可合成符合该通式的一大类衍生物,活性基团种类较多,能抑制金黄色葡萄球菌等多种菌株,并具有一定的光电性能,应用领域更为广阔.
猜你喜欢
分子催化(2022年1期)2022-11-02
农产品质量与安全(2022年4期)2022-08-24
医学概论(2022年4期)2022-04-24
今日农业(2021年2期)2021-11-27
科技与创新(2020年16期)2020-08-18
宇航材料工艺(2020年1期)2020-03-26
科技与创新(2018年2期)2018-01-09
今日农药(2017年2期)2017-03-24
农村农业农民·B版(2016年5期)2016-05-14
湖北农业科学(2014年9期)2014-08-08
