
过渡金属硫族二维材料吸附Li2Sn的理论研究
2022-08-23 11:28陈许敏胡旖航
杭州电子科技大学学报(自然科学版) 2022年4期
陈许敏,程 杰,胡旖航,张 科,叶 盼
(杭州电子科技大学理学院,浙江 杭州 310018)
0 引 言
目前,锂电池能提供的能量密度正接近阙值,寻找新的替代材料以提高电池性能显得极为重要[1]。锂硫电池能提供1 675 mAh/g的比容量和2 600 Wh/kg的比能量密度,一直备受关注[2]。相比于锂电池,锂硫电池的能量密度更高,且硫元素在自然界中分布广泛且价格低廉[3-4]。然而,锂硫电池也同样面临着多种挑战,如单质硫和硫化锂的低导电性使得电池能量利用率低、循环性能差[5];多硫化锂中间产物溶解于常用液体电解质时,触发“穿梭效应”,降低放电效率,导致电池中有效物质的不可逆损失,缩短电池的循环寿命[6-8]。因此,如果要降低多硫化锂在电解质中的溶解,使用的材料与多硫化物之间需要产生较强的吸附作用,并具有良好的导电性。过渡金属硫族化合物(Transition Metal Dichalcogenides,TMD)具有优良的电化学性质。Zhang等[9]研究发现,TiS2和NbS2等过渡金属硫化物具有削弱穿梭效应的作用。Tao等[10]通过密度泛函理论计算发现,相较于TiO2—S,Ti4O7—S材料具有更高的可逆容量和更好的循环性能。Yuan等[11]指出,混有CoS2的电极材料能够确保高放电容量和优秀的循环性能。因此,TMD是吸附锂离子的理想半导体材料。Pang等[12]运用第一性原理进行计算研究,合成得到Co9S8的性质,并将其涂在硫化物电极表面,使得电池循环更加稳定。田娇[13]采用过渡金属化合物和聚多巴胺(Polydopamine, PDA)的混合材料作为正极材料,研究锂硫电池的性质,实验结果表明,这种材料不仅能改善材料的整体性能,还对极性多硫化物有较强的吸附作用,但对该材料体系吸附性质的综合研究只限于理论层面,不够全面。相对于其它材料而言,二碲化钨(WTe2)的比表面积大,能提供给Li2Sn的吸附位点多,故本文选取WTe2及与其性质相似的二碲化钼(MoTe2)和二硒化钨(WSe2)这3种TMD,采用第一性原理的差分电荷密度等方法,研究WTe2,MoTe2和WSe2与锂硫化合物的作用机理,及其对体系的吸附性质。
1 计算方法与模型
本文使用基于密度泛函理论的计算软件包VASP(Vienna Ab-initio Simulation Package)[14-15]进行计算。其中,能量交换关联能函数采用Perdew-Burke-Ernzerhof(PBE)[16-17]形式的广义梯度近似(General Gradient Approximate,GGA)法。
结构优化和计算是采用截断能(cut-off energy)为400 eV的平面波基组展开,在布里渊区计算时采用VASP软件推荐的原点在Г点的Monkhorst-Pack型网格[18],结构使用3×3×1 Monkhorst-Pack K点,驰豫计算时核离子步收敛精度取为5 eV/nm,能量收敛精度取为1.0×10-4eV,采用DFT-D2色散校正方法[19-20]修正Li2Sn与吸附材料之间的范德华相互作用。
WTe2,MoTe2和WSe2都拥有与BN,MoS2等类石墨烯结构的二维层状材料类似的结构[21],结构优化后的WTe2,MoTe2和WSe2超晶胞的俯视图如图1所示。图1(a)中,W原子占据了六边形的一个亚晶格,Te原子占据了六边形的另一个亚晶格;图1(b)中,用同族的Mo原子替换了原有的Te原子;图1(c)中,用同族的Se原子替换了Te原子。本文的计算模型选取4×4×1的单层超胞结构,共32个原子,确保相邻分子之间的距离大于1.2 nm[22-23],避免了相邻吸附分子之间的相互作用。另外,真空层取1.5 nm厚度,避免了层间干扰。结构优化时,对所有原子进行驰豫。
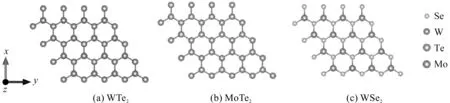
图1 3种过渡金属硫族化合物的俯视结构图
2 计算结果与分析
2.1 吸附能和吸附构型
为了得到较稳定的初始结构,在衬底吸附小分子前,分别对衬底及小分子进行结构优化,然后进行模拟计算,分析Li2Sn(n=1,2,4,6)与二维材料(WTe2,MoTe2,WSe2)吸附后体系的结构模型,用第一性原理计算吸附体系的结构优化模型,用于分析键长变化。
WTe2表面吸附Li2Sn(n=1,2,4,6)的结构优化结果如图2所示。从图2(a)—2(d)可以看出,随着吸附分子中S原子的不断增加,2个Li原子的间距逐渐减小。图2(a)中,2条Li—S键几乎平行于WTe2表面。Li2S吸附在WTe2表面后,2个Li原子沿着W—Te键往外移动。实际计算结果也表明,Li1原子和Li2原子分别偏移了0.015 6 nm和0.025 9 nm,而S原子沿着W—Te键往中心的谷位偏移了0.063 0 nm,同时,Li2S的键角由自由分子的109.471°升高至吸附后的150.495°,Te原子对Li原子的吸附使得键角增加。图2(b)中,Li2S2吸附在WTe2表面时,2个Li原子和顶位的Te原子基本重合,1个S原子与顶位的W原子基本重合,另1个S原子与晶胞的谷位基本重合。图2(d)中,Li2S6体系较大,吸附后材料的对称性较低,吸附作用显著降低。Li2Sn(n=1,2,4,6)在MoTe2和WSe2表面的稳定吸附构型结构与Li2Sn(n=1,2,4,6)在WTe2表面的稳定吸附构型相似,这里就不再分析。
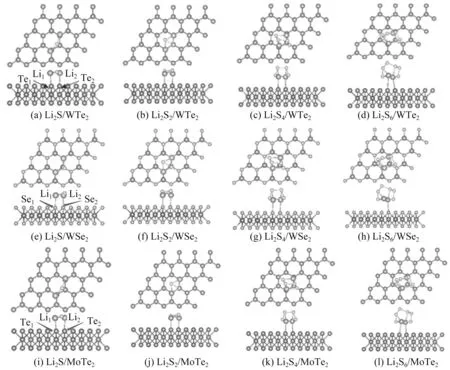
图2 Li2Sn(n=1,2,4,6)在3种二维材料表面的稳定吸附构型
为了分析Li2Sn分子在不同吸附材料上的稳定性,计算Li2Sn分子的吸附能Eads[24],
Eads=ELi2Sn/WTe2-EWTe2-ELi2Sn
(1)
式中,ELi2Sn/WTe2,EWTe2,ELi2Sn分别表示Li2Sn吸附WTe2表面后体系的总能量、吸附前WTe2的能量、吸附前Li2Sn的能量。Li2Sn分子吸附在MoTe2和WSe2表面的计算方法与式(1)类似。Eads为正值时,吸附过程从外界吸收能量;Eads为负值时,吸附过程放出能量,其绝对值越大,放出的热量越多,结构越稳定。分别计算Li2Sn(n=1,2,4,6)在WTe2,WSe2,MoTe2表面的吸附能,结果如表1所示。

表1 Li2Sn在WTe2,WSe2,MoTe2表面的吸附能 单位:eV
从表1可以看出,MoTe2表面吸附Li2S4小分子的体系具有最高的吸附能,为-1.83 eV,吸附过程中放出的能量最多,体系结构最稳定。
Li2Sn分子吸附二维材料表面时,有多个可能的吸附位置,Li2S分子中的S原子吸附在衬底不同位置。其中,S原子分别位于Te/Se上方的顶位1、W/Mo上方的顶位2、谷位和桥位。为了分析Li2Sn分子吸附位置对吸附能的影响,分别计算Li2S在3种二维材料表面不同位置的吸附能,结果如表2所示。
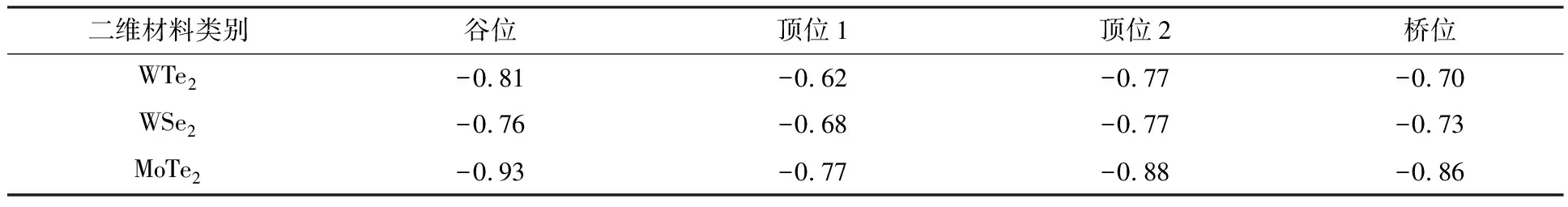
表2 Li2S在WTe2,WSe2,MoTe2表面不同位置的吸附能 单位:eV
从表2可以看出,在二维材料表面的不同吸附位置上,Li2S的吸附能存在差异,但最大吸附能所对应的吸附位置都在谷位。因此,通常情况下,谷位是小分子最稳定的吸附位置。对于同一谷位的吸附位置,3种二维材料中,MoTe2衬底的吸附能最大,WTe2衬底的吸附能次之,WSe2衬底的吸附能最小。
为了进一步分析Li—Te键和Li—Se键对吸附造成的影响,分别计算每种吸附构型对应化学键的键长,结果如表3所示。
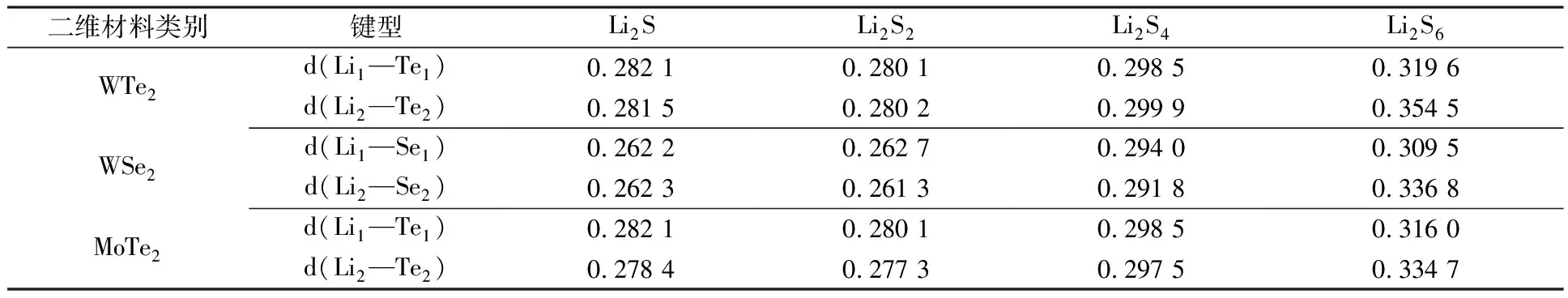
表3 二维材料吸附Li2Sn结构中的Li—Te/Se键长 单位:nm
从表3可以看出,Li2Sn吸附在WTe2表面时,随着锂硫化合物中硫元素的增加,整体上Li1—Te1的键长呈增大趋势,但是Li2S2的键长最短,是个特例。相较于几乎未发生偏移的Li2S2体系,Li2S体系中朝外偏移的Li原子与Li2S4体系中朝内偏移的Li原子都整体拉长了Li1—Te1键,Li2—Te2键长具有与Li1—Te1相似的变化规律。文献[25]指出,Li2Te晶相结构的Li—Te键长为0.281 6 nm,Li2Se晶相结构的Li—Se键长为0.260 0 nm。因此,0.281 6 nm附近的Li—Te键长和0.260 0 nm附近的Li—Se键长都意味着Li原子与Te/Se原子之间形成了较强的化学键,体系呈现化学吸附。因此,Li2Sn吸附在MoTe2表面与WTe2表面时,化学作用力大多由Li正离子和Te负离子之间的离子键提供。
2.2 差分电荷密度分析
为了进一步分析Li2Sn吸附在不同二维材料表面的电子相互作用,计算Li2Sn吸附在不同二维材料(WTe2,WSe2,MoTe2)表面前后的差分电荷密度,结果如图3所示,其中上半部分为俯视图,下半部分为侧视图。计算差分电荷密度的公式如下[26]:
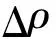
(2)
式中,ρLi2Sn/WTe2表示吸附体系的总电荷密度,ρLi2Sn和ρWTe2分别表示只吸附WTe2和Li2Sn的电荷密度,MoTe2和WSe2的计算方法与式(2)类似。
Li2Sn(n=1,2,4,6)吸附在WSe2表面的差分电荷密度如图3所示。
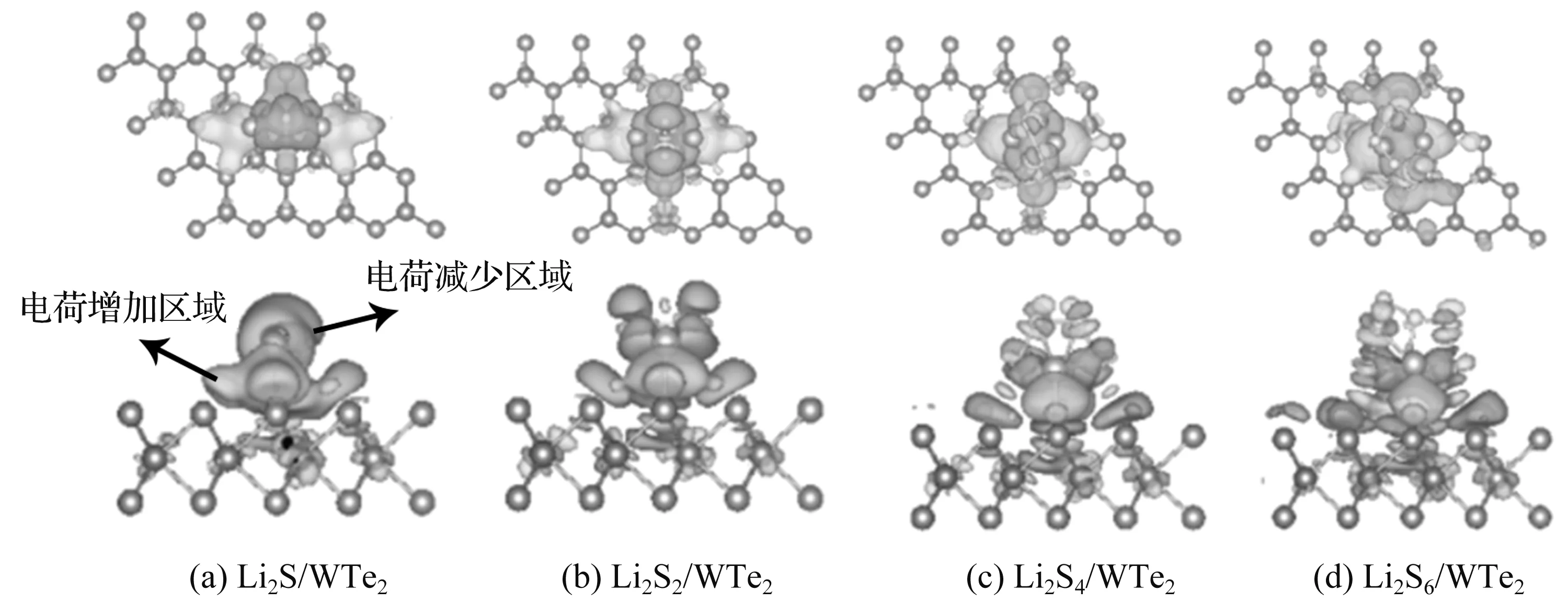
图3 锂硫化合物吸附在WTe2表面的差分电荷密度图
从图3(a)可以看出,当Li2S吸附在WTe2表面时,Li2S团簇的电荷分布发生转移,Li—S键区域的电荷密度减少,Li—Te键区域的电荷密度增加。从图3(b)可以看出,小分子替换成Li2S2后,电荷密度的变化与Li2S类似,WTe2吸附Li2S2体系的电荷密度减少区域主要沿Li2S2团簇中心上下对称分布,且电荷转移量减少,这种上下对称的分布与图3(a)中Li2S吸附体系的集中分布相比有明显的不同;在Li2S的基础上,增加一个S原子后,电荷密度减少区域不再集中于Li2Sn团簇,而是分布在上下两侧,电荷转移的程度较低,Li2Sn团簇体系对电荷转移的贡献减弱。随着S原子个数的增加,电荷密度减少区域逐渐分散,到WTe2吸附Li2S6体系时,从图3(d)可以看出,电荷密度减少区域大幅度分散,Li2S6在WTe2上的电荷密度变化显著下降,说明Li2S与WTe2之间的化学作用强于Li2S6,与吸附能有较好的关联性。
Li2Sn(n=1,2,4,6)吸附在WSe2表面的差分电荷密度如图4所示。
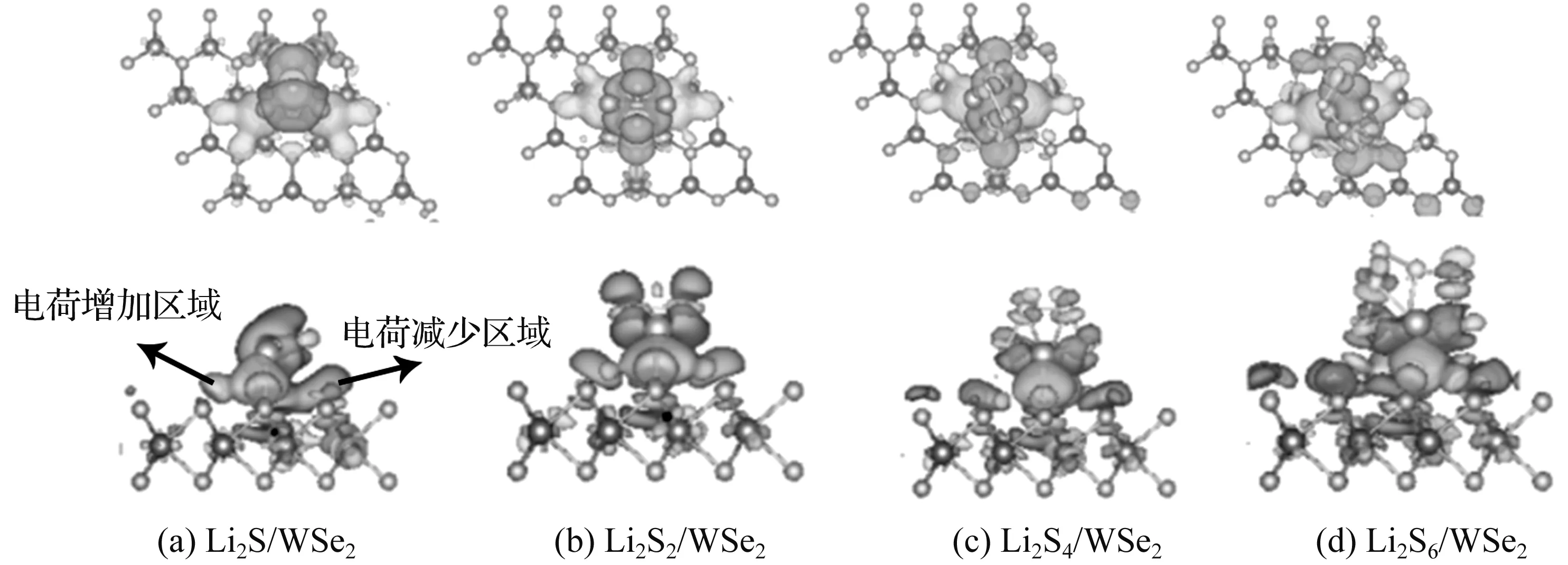
图4 锂硫化合物吸附在WSe2表面的差分电荷密度图
从图4可以看出,Li2Sn吸附在WSe2表面电荷的转移趋于横向扩散,吸附后的电子主要由Li2Sn转移到WSe2表面。不同的吸附构型上,在吸附界面处都聚集了大量的电荷密度,即Li2Sn与WSe2基底成键处的电荷密度是连续分布的。在吸附构型的根部,电荷密度聚集较多,而在吸附构型末端,电荷密度离散且趋于脱离状态。伴随S原子个数的增加,电子的离散化现象逐渐突出;当Li2S6吸附在WSe2衬底上时,对接触面的作用最强,但得失电子的区域局域化较低,导致Li原子与Se原子的相互作用减弱,造成Li原子与Se原子之间距离增大,这与表3的键长分析结果保持一致。
Li2Sn(n=1,2,4,6)吸附MoTe2的差分电荷密度如图5所示。
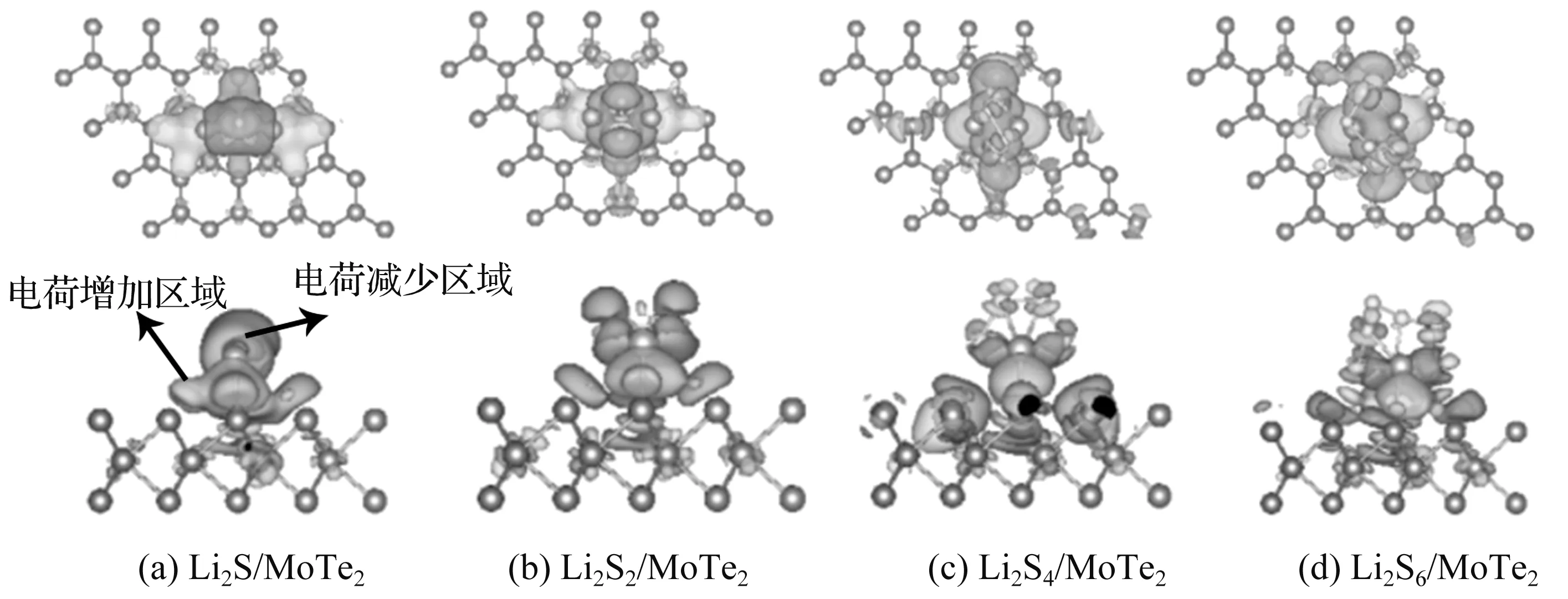
图5 锂硫化合物吸附在MoTe2表面的差分电荷密度图
从图5可以看出,Li2Sn吸附在MoTe2衬底时,随着S原子个数的增加,电荷转移的横向分布趋势逐渐减弱,与吸附在其它衬底相似,电子的区域局域化明显降低。Li2S和Li2S2吸附在MoTe2衬底上时,电子区域主要集中在S原子附近,得到电子区域主要集中在接触面附近,表明转移到界面附近的电子主要由S原子贡献。Li2S4和Li2S6吸附在MoTe2衬底时,电子密度的横向趋势减弱,表明Te与Li2S6的S有明显的电荷转移,电子的转移使Li—S键逐渐被打开,S原子远离Li原子,趋于形成S单质,最终实现Li—S的解离。
综上分析可以发现,Li2Sn吸附在不同过渡金属WTe2,WSe2和MoTe2表面时,吸附后Li2Sn小分子化合物的电荷密度均沿着Li—Te键轴方向增加,沿着Li—S键轴方向减少,但Li2Sn在MoTe2上的电荷密度变化比在WTe2和WSe2上更明显,说明作为Li2Sn的锚定材料,MoTe2有更强的吸附能力。
为了直观观察3种二维材料吸附Li2S后的电子重新分布状态,本文采用差分电荷密度切面图的形式进行展示,结果如图6所示。为了便于分析,本文淡化处理了背景。

图6 Li2S吸附在过渡金属硫化物表面的差分电荷密度切面图
从图6(a)可以看出,Li—Te键轴方向的电荷密度增加,而Li—S键轴方向的电荷密度减少,电荷密度变化表明:Li2S吸附到WTe2表面后,电子从Li2S转移到WTe2表面,Li2S和WTe2之间形成类离子键;电荷转移加强了Li原子与Te原子之间的作用,同时削弱了Li2S团簇内部的Li—S键,并且Li2S与衬底材料表面的Te/Se原子发生了显著的相互作用,从而引起了电子间的杂化及偏移。对于图6(b)的WSe2吸附Li2S体系和图6(c)的MoTe2吸附Li2S体系,与WTe2吸附Li2S体系类似,部分电荷从Li2S转移到WTe2,MoTe2。3种吸附衬底中,MoTe2作为衬底吸附Li2S时的Li原子与Te原子之间的差分电荷密度最大,S原子与Li原子之间的差分电荷密度最小,说明其电荷密度变化最明显,产生更强烈的相互作用,进一步说明MoTe2的吸附能是最大。
3 结束语
本文采用密度泛函理论的第一性原理进行模拟计算,系统研究了锂硫化合物在过渡金属硫族二维材料WTe2,MoTe2和WSe2上的吸附性质。研究发现,MoTe2具有较强的Li2Sn吸附能力;MoTe2与Li2Sn之间的电荷转移最多,结构最稳定;在过渡金属硫族二维材料表面吸附锂硫化合物的体系中,对吸附作用产生较大影响的因素主要是3种二维材料表面的碲或硒原子与锂硫化合物的锂原子产生的静电作用。本文的研究为寻找理想的锂硫电池电极锚定材料提供理论依据,为提高锂硫电池的循环稳定性提供新的解决思路。然而,仅用金属硫化物作为辅助材料仍不能有效解决硫固有的导电率较低等问题,必须通过其它方式提高硫的利用率,因此,寻求新型、可替代的硫电极材料是本文未来的研究重点。
猜你喜欢
上海师范大学学报·自然科学版(2022年3期)2022-07-11
科学导报(2022年29期)2022-05-26
小作家报·教研博览(2022年11期)2022-04-02
科学中国人·下旬刊(2021年12期)2021-03-10
河南科技(2020年6期)2020-10-21
华东师范大学学报(自然科学版)(2020年1期)2020-03-16
上海师范大学学报·自然科学版(2018年3期)2018-05-14
计算机应用(2016年10期)2017-05-12
新高考·高一物理(2016年7期)2017-01-23
新高考·高一物理(2015年6期)2015-09-28
