
利用单碱基编辑器定点突变猪肌肉生长抑制素基因的研究
2022-08-23 02:39毕延震
中国畜牧兽医 2022年8期
王 晶,朱 喆,张 鹏,毕延震
(1.中南民族大学生命科学学院,武汉 430074;2.湖北省农业科学院畜牧兽医研究所,动物胚胎工程与分子育种湖北重点实验室,武汉 430064)
中国猪种质资源丰富,占全世界1/3左右。宁乡花猪是中国本土著名品种,具有肌内脂肪含量高、繁殖力高、抗病抗逆性强等优势,但其生长速度、饲料报酬和瘦肉率低等缺点限制了该品种在市场中的发展。因此,如何在规避缺点的同时最大化利用其内在优势,对宁乡花猪乃至整个地方猪养殖行业发展有着重要意义。
传统猪遗传育种周期长且成本高。目前,以CRISPR/Cas9技术为基础的基因编辑技术已经广泛应用于一个或多个优良性状的研究,该方法在猪种质资源创新中具有重要意义[1-2]。在CRISPR/Cas9编辑系统中,sgRNA与Cas9蛋白形成的复合物会切割靶DNA,导致靶DNA双链断裂(double strand breaks,DSB),DSB是最严重的DNA损伤形式之一,可能会引起染色体结构异常或缺失,扰乱细胞正常功能,最终导致细胞死亡[3-4]。因此,如何能在不产生DSB的情况下进行精准编辑成为亟需解决的难题。2016年,以CRISPR/Cas9为基础的单碱基编辑技术应运而生,该技术在不造成DSB的情况下精准突变单个碱基,其原理是将失去切割活性的核酸酶Cas9蛋白(deactiaved Cas9,dCas9)和作用于单链DNA(single-stranded DNA,ssDNA)碱基的脱氨酶融合,依靠sgRNA将碱基编辑酶锚定到靶位点上,从而使局部ssDNA上的胞嘧啶(cytosine,C)或腺嘌呤(adenine,A)脱氨,实现精准C→T或A→G替换[5],其中,胞嘧啶碱基编辑器(cytosine base editor,CBE)可以将密码子(CAA、CAG、CGA或TGG)精准转换成终止密码子(TAG、TAA或TGA)来阻止翻译[6]。与传统CRISPR/Cas9技术相比,单碱基编辑器能在避免发生DSB及碱基缺失或增加的情况下实现基因突变,减少DNA损伤和细胞死亡[5]。近年来,科学家致力于改进碱基编辑器,已经产生了具有高编辑效率[7]、窄编辑窗口和广泛前间隔序列邻近基序(protospacer adjacent motif,PAM)识别位点的多种变体[8],且应用于多种动物基因编辑[9-11]。
肌肉生长抑制素(myostatin,MSTN)又称生长分化因子8,主要在骨骼肌中表达,对肌肉发育和再生具有负调控作用[12]。MSTN基因自然突变会使牛表现出“双肌”性状[12],而宁乡花猪在自然情况下不存在MSTN基因有益突变。研究发现,人为敲除MSTN基因会导致猪背膘厚度下降,脂肪含量减少[13],证明了运用基因编辑的方法进行遗传改良是有效的。因此,本研究利用单碱基编辑技术编辑宁乡花猪的MSTN基因,利用CBE在基因编码区定向引入终止密码子,建立基因敲除的单碱基编辑体系,为培育MSTN碱基编辑猪奠定基础。
1 材料与方法
1.1 材料
宁乡花猪来源于湖南楚沩香农牧股份有限公司保种场。单碱基编辑器YE1-BE3-FNLS由中国农业科学院赠送。pMLM3636-puro由湖北省农业科学院畜牧兽医研究所合成保存;DNA Ladder和Premix酶均购自TaKaRa公司;限制性内切酶购自Thermo Scientific公司;嘌呤霉素由Biosharp提供;DMEM和DPBS均购自HyClone公司;血清购自Invigentech公司。
1.2 方法
1.2.1 宁乡花猪肾原代成纤维细胞的培养 取7日龄宁乡花猪,用静脉注射法致死,75%乙醇溶液消毒全身,酒精棉球擦拭腹部后,用手术刀割开猪腹部,取出肾组织,用生理盐水清洗5~6次,再用含1%双抗(青霉素和链霉素混合溶液)的DPBS清洗,将组织放入培养皿中,剪去表面膜,将肾组织剪成约1 mm×1 mm×1 mm大小的组织块,用枪头吸取将组织块均匀铺在6孔板中。于5% CO2、37 ℃培养箱倒置1 h后,加入2 mL完全培养基(DMEM+10%FBS+1%双抗),继续放入培养箱中培养,每隔2 d换一次液,5~7 d后去除组织块。
1.2.2 sgRNA及检测引物设计 在宁乡花猪MSTN基因第2外显子(Chromosome 15,GeneID:399534)处设计sgRNA。sgRNA的设计满足以下条件:①sgRNA的长度为20 bp,PAM序列为NGG;②远离PAM位点的4—8 bp处,正义链需要有C,反义链需要有G;③转变后的碱基(C→T或G→A)与相邻碱基可以将原有氨基酸转变成终止密码子(TAA、TAG、TGA);④sgRNA的GC含量在40%~60%之间。以此为基础,设计sgRNA(表1),并在正义链的5′-端添加ACACC酶切位点,反义链5′-端添加AAAAC保护碱基。根据MSTN基因序列,运用Primer Premier 3.0软件设计MSTN基因扩增序列引物(表1)。引物由生工生物工程(上海)股份有限公司合成。

表1 引物信息
1.2.3 sgRNA表达载体构建 将sgRNA插入多克隆位点5′-BsmBI-3′之间,pMLM3636-puro质粒经内切酶BsmBⅠ酶切后,用胶回收试剂盒回收切割成功的酶切产物。将合成的sgRNA稀释成10 μmol/L后退火形成双链,反应体系20 μL:sgRNA-F 1 μL,sgRNA-R 1 μL,10×NEB Buffer 2 μL,用ddH2O补足至20 μL。反应条件为:95 ℃,5 min;37 ℃,1 h;4 ℃保存。将线性化的pMLM3636-puro载体与双链sgRNA连接,反应体系10 μL:线性化pMLM3636-puro 1 μL,sgRNA 3 μL,Solution Ⅰ 5 μL,ddH2O补足至10 μL。置于37 ℃恒温水浴锅中连接5 h,构建PMLM3636-puro-MSTN;将连接好的载体转化大肠杆菌DH5α感受态细胞,活化后均匀涂布在含有氨苄青霉素抗性的LB培养皿上,37 ℃倒置过夜培养,挑取单菌落,摇菌后将菌液送生工生物工程(上海)股份有限公司测序验证;利用无内毒素质粒小提中量试剂盒提取质粒,通过凝胶电泳判断pMLM3636-puro和pMLM3636-puro-MSTN大小。
1.2.4 电转液配制 配制10×电转液:KCl 4.473 g,CaCl20.0083425 g,Hepes 2.9787 g,EDTA 0.37224 g,无水MgCl20.238 g,K2HPO41.14 g,调节pH为7.6,加ddH2O定容至50 mL,于4 ℃保存。使用前用ddH2O稀释为1×电转液,0.22 μm滤器过滤,分装后,4 ℃保存备用。
1.2.5 电转染宁乡花猪肾成纤维细胞 将宁乡花猪肾成纤维细胞复苏并置于6孔板中培养,当细胞汇合度达到90%时,用0.25%胰酶消化细胞,1 min后,加入适量含10% FBS的DMEM培养基,吹打细胞,至细胞分散,收集细胞至1.5 mL离心管中;1 000 r/min离心2 min,弃去上清液,依次用DPBS和电转液吹打洗涤,离心后弃去上清液,将提前准备好的质粒(YE1-BE3-FNLS:6 μg,pMLM3636-puro-MSTN:3 μg)、细胞沉淀和电转液共100 μL转入电转杯中,电转条件:220 V,3 ms,1 pulse;电转完成后将电转杯中的液体全部吸取接种于100 mm的培养皿中,37 ℃、5% CO2培养箱中培养,24 h后更换新鲜DMEM培养基,同时加入嘌呤霉素进行药筛,并观察荧光表达情况。利用Image J软件计算不同视野下带有红色荧光的细胞占该视野下所有细胞的比例,并计算转染效率(整体平均值)。
1.2.6 单克隆细胞挑取和Sanger测序分析 药筛72 h后,更换新鲜含10% FBS的DMEM培养基,每3 d换一次液,培养7 d后,挑取单克隆细胞至48孔板,待48孔板长满后,传代至24孔板,分别取200 μL单克隆细胞和野生型细胞悬液,用细胞裂解液裂解后直接作为PCR模板,使用检测引物扩增。PCR反应体系25 μL:2×PremixTaq12.5 μL,DNA模板 1 μL,MSTN-F 1 μL,MSTN-R 1 μL,补充ddH2O至25 μL。PCR反应条件:94 ℃预变性5 min;94 ℃变性30 s,59 ℃退火30 min,72 ℃延伸1 min,共32个循环;72 ℃终延伸7 min。用1.5%琼脂糖凝胶电泳检测,将鉴定正确的PCR产物送至生工生物工程(上海)股份有限公司测序。
1.2.7 Western blotting检测MSTN蛋白的表达情况 将野生型细胞和阳性单克隆细胞进行扩大培养,收集细胞,离心后加入蛋白裂解液,冰浴30 min后,4 ℃、12 000 r/min离心15 min,收集上清液。测定蛋白浓度后按比例加入适量SDS和RIPA混匀,95 ℃ 10 min,蛋白变性后,-20 ℃保存备用。在120 V、1.5 h条件下进行SDS-PAGE分离蛋白,聚丙烯酰胺凝胶浓度为10%,电泳后将蛋白转移至PVDF固态膜上,再经5%脱脂奶粉常温封闭2 h,一抗孵育2 h,二抗孵育1 h,最后滴加显色液后显影。采用Image J软件对Western blotting结果进行量化分析,以β-tubulin作为内参对照。试验数据用GraphPad Prism 9.0进行统计学分析,结果以平均值±标准差表示,采用t检验分析差异显著性,以P<0.01表示差异极显著。
2 结 果
2.1 宁乡花猪肾原代成纤维细胞的培养
采用组织贴壁法分离宁乡花猪肾原代成纤维细胞。培养3 d后可以明显观察组织块周围有细胞生长,细胞以纺锤形为主,形态不均一,生长旺盛,培养7 d后,除去组织块,将剩余细胞传代至6孔板中,2 d后更换培养基,继续培养,细胞呈现梭形,边缘清晰,形态均一,生长力旺盛(图1),说明成功分离获得宁乡花猪肾原代成纤维细胞。

A,原代培养;B,F2传代培养A,Primary culture;B,F2 subculture图1 宁乡花猪肾成纤维细胞形态(100×)Fig.1 Morphology of kidney fibroblasts in Ningxiang pig (100×)
2.2 构建sgRNA表达载体并转染宁乡花猪肾成纤维细胞
测序结果显示,pMLM3636-puro-MSTN质粒构建成功(图2)。采用电转染的方法将单碱基编辑器YE1-BE3-FNLS和重组表达载体pMLM3636-puro-MSTN共转染至肾成纤维细胞,24 h后观察红色荧光表达情况(图3),利用Image J软件计算转染效率为67.4%。
2.3 阳性单克隆细胞筛选和基因靶位点测序结果分析
对挑取至24孔板中培养的单克隆细胞进行PCR鉴定。结果显示,MSTN基因PCR扩增产物大小为346 bp(图4A)。Sanger测序结果表明,55个单克隆细胞中,有3个阳性单克隆细胞在目标位点发生突变,目标位点突变率为5.5%(3/55)(图4A),其中仅10号阳性单克隆细胞在目标位点附近无脱靶现象。另外,有9个在非目标位点发生突变,非目标位点突变率为16.4%(9/55)(图4B),其中非目标突变位点在第3位,并不在传统的碱基编辑窗口(4—8 bp)之内。
2.4 Western blotting检测MSTN蛋白的表达
为进一步确定MSTN基因敲除的可靠性,选择仅含目标突变的10号阳性单克隆细胞进行Western blotting检测。由图5可知,以β-tubulin为内参,与野生型相比,试验组10号单克隆细胞MSTN蛋白表达量下降约60%。说明在MSTN第2外显子产生的终止密码子能够有效降低MSTN蛋白水平。

M,15000 bp DNA Marker;1,pMLM3636-puro;2,pMLM3636-puro-MSTN图2 pMLM3636-puro-MSTN质粒的电泳图(A)和测序峰图(B)Fig.2 Electropherogram (A) and sequencing map (B) of pMLM3636-puro-MSTN plasmid
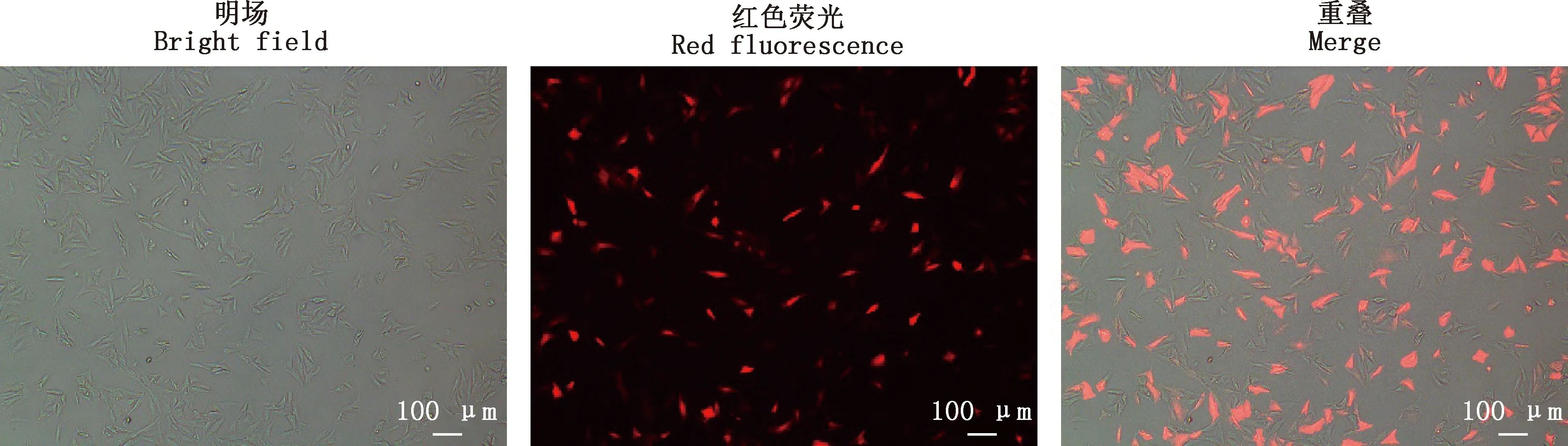
图3 电转染24 h后红色荧光表达情况(100×)Fig.3 Expression of red fluorescence after electransfection for 24 h (100×)
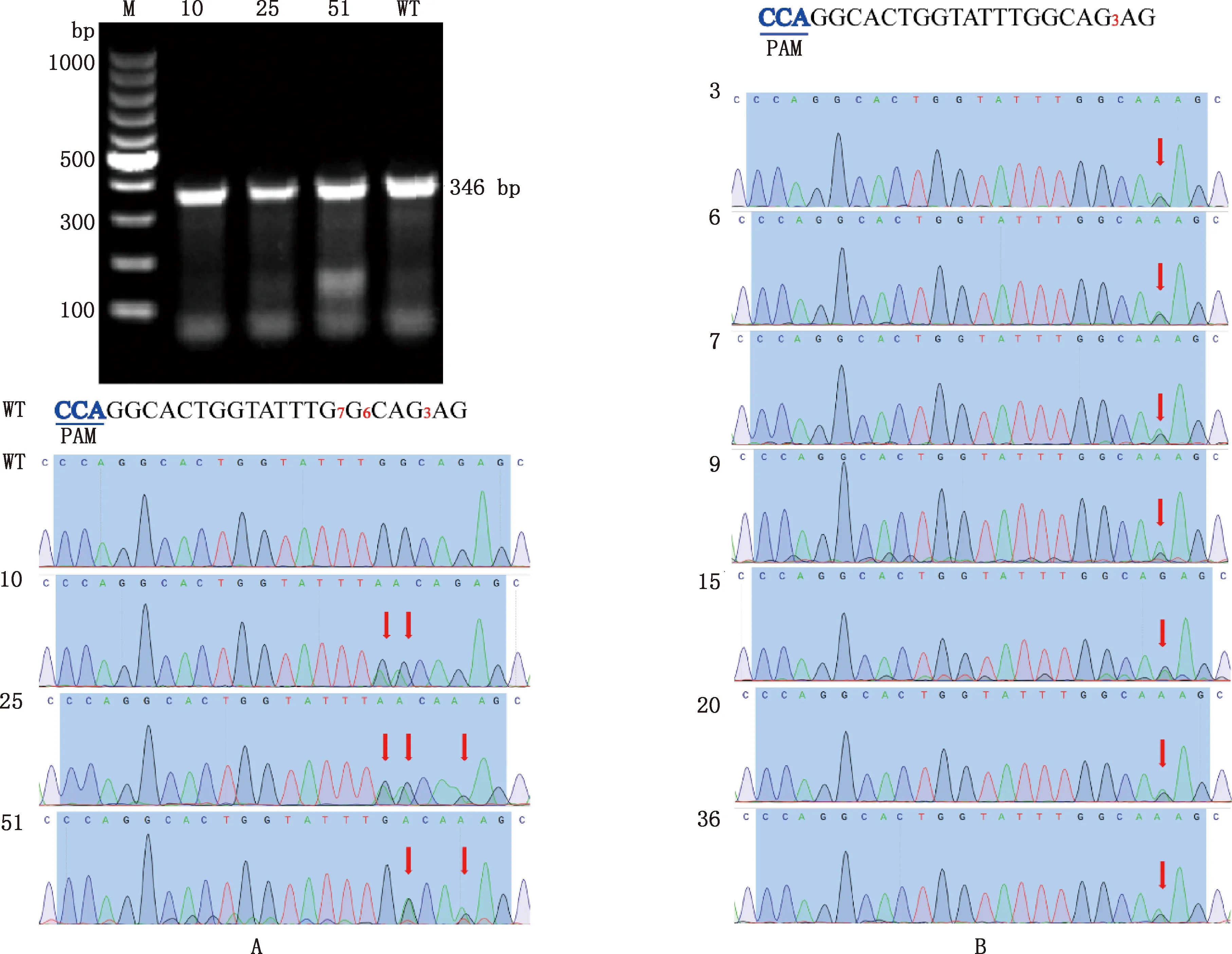
①A,目标突变单克隆细胞电泳图和测序峰图;B,非目标突变单克隆细胞测序峰图。②M,100 bp DNA Ladder;WT,野生型;数字10、25、51、3、6、7、9、15、20和36,突变单克隆细胞编号①A,Electrophoretogram and sequencing map of target mutant monoclonal cells;B,Sequencing map of non-target mutant monoclonal cells.②M,100 bp DNA Ladder;WT,Wild type;Numbers 10,25,51,3,6,7,9,15,20 and 36,The mutant monoclonal cell number,respectively图4 靶位点测序峰图Fig.4 Sequencing map of target sites

①WT,野生型;10,10号单克隆细胞。②**,差异极显著(P<0.01)①WT,Wild type;10,No.10 single clone.②**,Extremely significant difference (P<0.01)图5 Western blotting检测MSTN蛋白表达量Fig.5 MSTN protein expression levels detected by Western blotting
3 讨 论
CRISPR/Cas9基因编辑技术目前已被广泛应用于畜禽的改良育种方面,但该技术也面临着一些问题,Cas9蛋白可以在不依赖sgRNA的情况下突变基因组,引起DSB的产生,导致基因组不稳定;其次,CRISPR/Cas9技术导致DSB而诱发的同源重组修复途径需要同源模板的参与才能实现碱基的精准替换,而如何选择供体模板形式和递送方式是目前存在的难题[14]。2016年,Komor等[5]利用dCas9蛋白,结合脱氨酶开发了单碱基编辑器,该工具能在避免双链DNA断裂且无需供体DNA的情况下实现对基因组DNA进行精确的点突变,具有更高的精确度和安全性。
本研究使用单碱基编辑器YE1-BE3-FNLS在MSTN基因第2外显子上人为引入终止密码子。目标位点G(C)→A(T)为C6和C7,编辑效率为5.5%。研究发现,利用“all-in-one”修饰的CBE得到的1个使MSTN基因提前终止的巴马猪的单克隆细胞系,编辑效率为5.6%[15],与本试验结果相近。根据文献报道,利用YE1-BE3-FNLS编辑HEK 293T细胞,在C5~C7位点的平均编辑效率为78.3%[16]。因此,将碱基编辑器用于编辑哺乳动物体细胞时,应该注意细胞类型对编辑效率的影响。
除此之外,在目标位点附近发现了脱靶现象,C3位点发现了G(C)→A(T)突变,编辑效率为16.4%,高于目标位点。推测可能与目标突变位点的序列有关。本研究目标突变位点的序列为5′-C(G)-GG(CC)-T(A)-3′(CC为突变位点),C之前的碱基为G,而非目标突变位点的序列为5′-A(T)-G(C)-A(T)-3′,且C之前碱基为T,该编辑器使用的脱氨酶是rAPOBEC1,其脱氨酶脱氨效率为TC>CC>GC[5],这解释了C3的编辑效率大于目标位点C6和C7的原因。因此,在设计sgRNA时应该注意目标位点附近的碱基序列,减少脱靶现象发生。研究证明,腺嘌呤碱基编辑器(adenine base editor,ABE)的特异性高于CBE[17],Jeong等[18]通过改造ABE得到的ABE7.10(P48R)变体,发现其具有更高的脱氨率和较低的腺嘌呤编辑率,它还可以实现TC→TT或TC→TG的替换,因此,利用ABE引入终止密码子是降低脱靶率的潜在方法之一。综上所述,针对CBE碱基编辑器在体细胞中编辑效率较低且在高C位点易脱靶的问题,笔者认为,鉴于ABE比CBE具有更高编辑效率和较低的脱靶率,因此可以利用ABE碱基编辑器人工创造终止密码子。
4 结 论
本研究利用电转染的方法将pMLM3636-puro-MSTN和YE1-BE3-FNLS共转入宁乡花猪的肾成纤维细胞中,在红色荧光和嘌呤霉素双筛选的情况下,获得了阳性单克隆细胞。结果显示,在MSTN基因第2外显子处成功引入终止密码子,且目标位点G→A的编辑效率为5.5%。与野生型相比,试验组10号单克隆细胞MSTN蛋白的表达量降低约60%。本研究成功运用了单碱基编辑技术在宁乡花猪MSTN基因编码区实现定点编辑,为后期生产瘦肉率高的碱基突变猪奠定基础。
猜你喜欢
科学之谜(2021年2期)2021-04-25
电脑爱好者(2021年3期)2021-02-06
科学导报(2020年54期)2020-09-09
小康(2020年3期)2020-02-10
作文评点报·低幼版(2019年42期)2019-12-30
计算机应用文摘·触控(2019年19期)2019-11-05
江苏科技报·E教中国(2019年2期)2019-09-10
学苑创造·B版(2019年5期)2019-06-14
科学24小时(2019年5期)2019-06-11
党的生活·党员电教与远程教育(2019年1期)2019-03-06
