
接触蛋白1与恶性肿瘤
2022-08-19 06:55张昊萌综述李长忠审校
济宁医学院学报 2022年4期
张昊萌 综述 李长忠,2△ 审校
(1山东大学附属省立医院;2山东第一医科大学附属省立医院,济南 250021)
癌症是死亡率最高的疾病之一,严重阻碍了预期寿命的延长和生活质量的提高。据世界卫生组织统计,2019年死于癌症的患者人数已超过心脑血管疾病。目前,已发现多种参与肿瘤生长、增殖、浸润、转移的通路及相关因子。接触蛋白1(contactin-1,CNTN-1)作为一种广泛参与细胞周期、微血管生成、淋巴管增殖、上皮-间充质转化等途径的蛋白,在恶性肿瘤发病中起到重要作用,也为未来治疗方案的研发及选择提供新的思路。
1 CNTN-1结构和功能
CNTN-1是免疫球蛋白(Ig)超家族contactin亚群,是一种糖基化磷脂酰肌醇(GPI)-锚定的神经元膜蛋白,在人脑和神经组织中大量存在。最早是在测定植物血凝素结合糖蛋白Gp135的氨基酸序列时发现,主要存在于细胞浆中[1-3]。CNTN-1蛋白上游基因定位在染色体12q11-q12[4],分子量约135KDa,主要由6个免疫球蛋白结构域,4个Ⅲ型纤维连接蛋白样(fibronectintype Ⅲ-like,FN Ⅲ-like)片段以及GPI构成[5]。
CNTN-1蛋白最初被认为是一种神经细胞黏附因子,可以与多种细胞表面蛋白相互作用,如,Ig超家族中的L1家族(L1、NrCAM 和神经肌成束蛋白)、RPTPα、RPTPβ、Notch受体等,并通过多种信号通路发挥调节细胞功能的作用。研究多数集中在神经系统方向,其主要功能包括促进神经元轴突生长,调节神经元纤维生长方向;在大脑发育的早期,脑室下区的神经前体细胞增殖依赖于CNTN-1蛋白的调控;也可能在神经细胞的增殖、分化的细胞骨架重塑过程中起到重要作用[6];CNTN-1蛋白的表达水平可能也与疼痛相关[7]。
2 CNTN-1蛋白与恶性肿瘤
2.1 CNTN-1在恶性肿瘤中的表达情况
CNTN-1蛋白作为一种参与肿瘤形成过程的相关调节蛋白,在多种人类肿瘤细胞中存在过表达,如肺腺癌、乳腺癌、食管癌、肝癌、胃癌、甲状腺癌、宫颈鳞癌等,并有相关证据提示可能与预后及疾病转归相关[8]
CNTN-1蛋白在正常组织、癌周正常组织及癌组织中表现出不同的表达水平,CNTN-1蛋白在多种组织来源的恶性肿瘤中高表达,提示CNTN-1蛋白可能为癌组织的特征表达产物,在肿瘤的疾病进展中发挥重要作用。见表1。
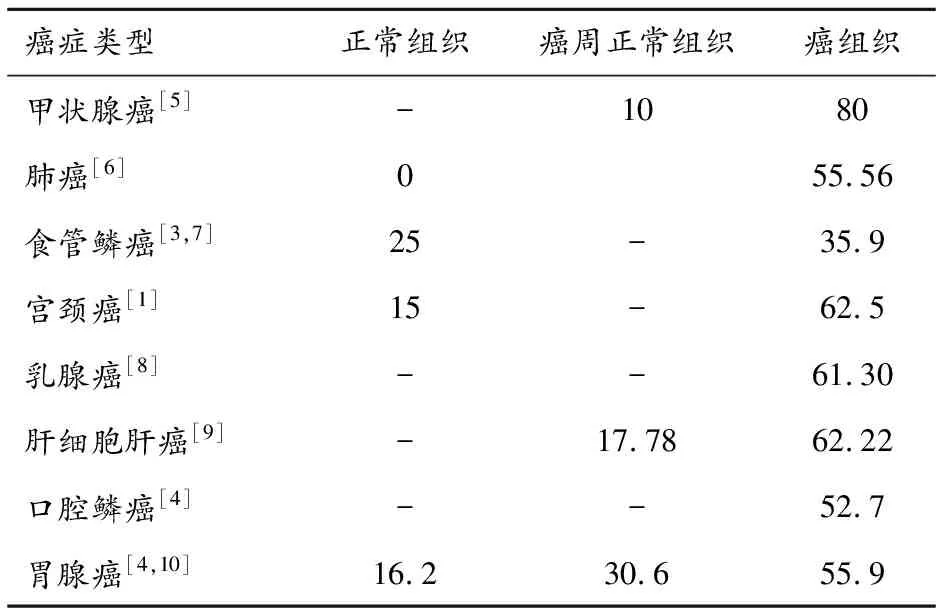
表1 CNTN-1在正常组织、癌周正常组织和癌组织中的表达阳性率(%)
2.2 CNTN-1蛋白促进肿瘤生长和转移的作用机制
2.2.1调节细胞周期 细胞周期调控是一系列复杂的机制,涉及多种细胞周期蛋白、细胞周期蛋白依赖性激酶、细胞周期检查点和细胞周期信号通路的调控[9]。其中一种或多种调控机制发生异常,则有可能出现细胞增殖的不可控,进而导致细胞癌变。CNTN-1蛋白在近期多项研究中被证实通过多种机制调节细胞周期,通过上调或下调CNTN-1的表达量,起到促进细胞周期或抑制细胞周期的作用[10-12]。
在肿瘤细胞株中上调CNTN-1蛋白的表达可以增强肿瘤细胞的增殖能力以及迁移侵袭能力,而降低其表达,癌细胞株的增殖速度、集落形成能力明显下降,且在基因沉默组中可观察到肿瘤细胞出现G1期阻滞的现象[10]。说明CNTN-1蛋白可能通过某些机制促进稳定细胞向不稳定细胞转化,促进稳定细胞由G0期进入G1期;也可能通过加速G1期细胞RNA复制、核糖体合成速度缩短细胞周期,实现加快肿瘤细胞增殖。目前,相关作用机制尚未发现和证实。
恶性程度更高、远处转移率更高、预后更差的三阴乳腺癌中CNTN-1蛋白的表达阳性率远高于非三阴乳腺癌。三阴乳腺癌中CNTN-1的表达与 Ki-67表达有一定的相关性[11]。Ki-67是一种位于细胞核、与细胞周期密切相关的因子,特点是半衰期较短,是常见表示细胞增殖能力与凋亡的指标,与肿瘤恶性程度成正相关[11]。Ki-67在G1、S、G2、M期均表达,而CNTN-1蛋白主要在G1期发挥作用,虽然现在已有证据不足以证实且明确Ki-67与CNTN-1蛋白的相互作用和调节机制,但可以推测Ki-67与CNTN-1蛋白在肿瘤发生过程具有协同作用,且CNTN-1蛋白高表达可能与肿瘤侵袭性高、淋巴结转移、预后差密切相关。
另在甲状腺癌的相关研究发现CNTN-1蛋白发挥作用,可能是通过RET/PTC3(Ret proto-oncogen Ret-activating protein ELE1)重组基因实现的,如果敲除CNTN-1蛋白上游基因可以有效抑制肿瘤细胞的增殖、侵袭,并且抑制细胞周期蛋白D1(Cyclin D1,CCND1)的表达[12]。通过影响Cylcin D1的表达,CNTN-1蛋白可调节肿瘤细胞生长周期,进而促进肿瘤细胞的增殖和生长[12]。
因此,CNTN-1蛋白在促进稳定细胞向不稳定细胞转化、在G0-S期多环节调整细胞生长周期、促进不稳定细胞增殖来实现肿瘤细胞的快速扩增,并通过与Ki-67协同表达进一步实现肿瘤细胞对周围组织的侵袭和转移。在未来的机制研究及相关药物研发中,存在通过特异性阻滞CNTN-1蛋白的表达阻滞肿瘤组织细胞周期运转来实现治疗的可能。
2.2.2调节VEGF-C相关基因表达 VEGF是一种具有重要的促血管生成活性的生长因子,对内皮细胞具有促进有丝分裂和抗凋亡、增加血管通透性、促进细胞迁移等作用,与实体肿瘤的生长、侵袭和转移密切相关[13]。
CNTN-1的表达与VEGF-C、HIF-1α的表达存在相关性[8]。CNTN-1可能是肿瘤细胞VEGF-C/FIT-4机制的下游效应因子之一。部分快速增殖的肿瘤细胞可能存在由于血供不足导致的缺氧,缺氧会诱导HIF-1ɑ因子地过表达,进而诱导VEGF-C表达提高以形成肿瘤供氧、提供营养物质的微小血管[8]。VEGF-C也可通过激活Scr-p38 MAPK 介导的C/EBP依赖信号通路的表达来增加CNTN-1的转录活性进而影响肿瘤细胞的生长和转移[14]。CNTN-1上游基因上有C/EBP的结合位点,在VEGF-C表达增加后,C/EBP与CNTN-1启动子的相对结合量显著增高[14],提高了CNTN-1蛋白的表达,继而增强了肿瘤细胞的转移性和侵袭性[15]。
CNTN-1表达增加也可促进VEGF-C的表达,进而引起VEGF-C与VEGFR-3相对结合量的增加,多种肿瘤的增殖、生长、浸润和淋巴结转移情况和VEGF-C/VEGFR-3基因过表达存在相关性[16]。VEGF-C通过ERK1/2和AKT信号通路,从而触发丝裂原活化蛋白激酶级联反应(mitogen-activated protein kinase,MAKP),刺激淋巴管内皮细胞增生;也可通过VEGFR-3酪氨酸残基Y1063发生磷酸化,诱导募集半胱氨酸的类受体蛋白激酶(cysteine-richreceptor-like kinase,CRK)Ⅰ/Ⅱ,使 c-Jun 氨基末端激酶 1/2(c-Jun N-terminal kinases1/2,JNK1/2)活化,促使毛细淋巴管内皮细胞分裂、增殖,从而促进新生淋巴窦和淋巴管的生成,提高肿瘤周围淋巴管的密度,进而促进肿瘤细胞通过淋巴道转移[17]。VEGF-C/Flt-4 诱导的 CNTN-1 表达提高,通过与细胞膜上的小GTP结合蛋白RhoA作用介导细胞内丝状肌动蛋白重排,并在迁移细胞中不断解聚和聚合,增强细胞侵袭,对细胞迁移运动至关重要[18]。
CNTN-1蛋白促进肿瘤转移的作用也可通过VEGF-C/VEGFR-3通路激活Notch/丝氨酸/苏氨酸蛋白激酶(serine/threonine protein kinose,AKT)信号通路实现,可抑制上皮性钙黏蛋白(E-cadherin)上游基因的转录,并且诱导E-钙黏蛋白与β-catenin复合物发生分解,破坏细胞间黏附、降低淋巴管壁的屏障功能、增加其通透性,提高肿瘤细胞通过淋巴系统的转移率[17]。
CNTN-1蛋白过表达作为VEGF-C相关通路发挥作用的促进因素可能是某些恶性肿瘤通过淋巴道转移的重要原因之一,在今后恶性肿瘤治疗新方案的研究中可能提供一个新的检测和治疗靶点。同时,通过靶向调控CNTN-1蛋白的表达,进而起到调控VEGF-C的作用,为治疗癌症术后淋巴转移和淋巴水肿的患者提供新的治疗策略。
2.2.3调节MMP-2和MMP-9表达水平 MMP在肿瘤转移过程中具有重要作用,通过降解细胞外基质促进肿瘤细胞浸润和侵袭。MMP的表达水平可受到CNTN-1蛋白地过表达和抑制的影响,CNTN-1过表达可促进MMP-2和MMP-9的表达,而CNTN-1上游基因的表达抑制可有效降低MMP-2和MMP-9的表达,因此,通过增加或者降低细胞外基质的降解可影响肿瘤细胞侵袭和迁移能力[19]。目前,CNTN-1调节MMP-2和MMP-9表达的具体分子机制尚不明确。
2.2.4影响上皮细胞-间质转化过程 上皮细胞-间充质转化(epithalial-mesenchymal transition,EMT)是指上皮细胞通过特定的程序转化为具有间质细胞表型的生物学过程[20]。在病理状态下,肿瘤细胞可以重新激活EMT程序,通过EMT,上皮细胞失去了细胞极性,失去了与基底膜的连接等上皮表型,获得了较高的迁移与侵袭、抗凋亡和降解细胞外基质等能力的间质表型。因此,EMT被认为是癌症进展和转移过程中的关键机制,并导致抗肿瘤药物耐药性。
EMT参与某些耐药癌细胞中肿瘤干细胞表型的形成,参与PI3K/Akt信号通路,也参与癌细胞产生对多西他赛的耐药性[21]。在此过程中,CNTN-1也发挥了重要的调节作用。在研究前列腺癌对多西他赛耐药性机制的实验中[22],发现通过降低抗多西他赛前列腺癌细胞中CNTN-1蛋白的表达,可以显著增加前列腺肿瘤细胞对多西他赛的药物敏感,并增加肿瘤细胞凋亡率。并且,可以在CNTN-1蛋白高表达组观察到与EMT有关的形态学变化。CNTN-1蛋白上游基因的抑制降低了间充质标志物的表达水平,但会增加上皮细胞相关标志物的表达。因此,敲除CNTN-1基因会阻碍抗多西他赛的前列腺癌细胞的增殖和EMT过程,起到抑制肿瘤增殖的作用。
在肺腺癌的相关实验中发现,CNTN-1重塑细胞肌动蛋白的骨架以及调节局部粘连结构的功能对于癌细胞的转移和浸润过程是必需的[23]。与EMT过程相关的肿瘤细胞的药物抵抗作用可能是通过细胞迁移能力的增加、获得肿瘤干细胞表型和细胞凋亡抵抗来实现的[23]。在既往的相关研究中发现,CNTN-1蛋白在药物抵抗细胞中的表达高于肿瘤祖细胞,说明肿瘤干细胞有很高的迁移能力和抵抗凋亡的能力,也可说明,抑制CNTN-1上游基因的表达可以增强癌细胞对化疗药物的敏感性,促进由铂类药物引起的肿瘤凋亡过程,进而引起对肿瘤细胞侵袭和转移的阻挡[24],实现化疗药物对肿瘤病变的治疗效果。
CNTN-1蛋白可能通过EMT过程在肿瘤进展中发挥作用,肿瘤细胞的淋巴结转移则是CNTN-1过表达的独立危险因素[25]。相关研究认为,CNTN-1发挥上述功能主要通过作为VEGF-C/FLt-4轴的下游效应分子,通过抑制PHLPP2信号介导的AKT去磷酸化降解E-钙粘着蛋白过程实现的[26]。这一传导通路可以影响EMT过程,进而促进肿瘤细胞增殖、浸润过程,可能与肿瘤细胞与顺铂的耐药性有关。另有研究认为CNTN-1蛋白通过RET/PTC3(Ret原癌基因和Ret-激活蛋白ELE1)重排基因调节来发挥作用的[27]。CNTN-1蛋白可能是EMT过程中最重要的黏附变化分子之一,是EMT过程重要的生物标志物和未来治疗靶点。
3 CNTN-1蛋白水平与多种肿瘤的预后关系
多项临床实验证实,CNTN-1蛋白的表达水平与患者预后相关,低表达患者的无瘤生存期、总生存期、3年及5年生存率均超过高表达患者,且存在统计学差异[8,28-29]。这提示CNTN-1蛋白的表达水平可能和患者的预后相关,并提供了作为观测指标检测疾病进展的指标的可能。
在宫颈癌患者中,CNTN-1蛋白低表达组的无瘤生存期约为(39.842±2.152)个月,而高表达组患者仅为(30.759±1.693)个月,明显低于低表达组,两组患者无瘤生存期的差异存在统计学差异[8]。
在肝细胞肝癌的患者中,CNTN-1高表达组患者的总生存期约为(19.96±9.39)个月,低于低表达组患者的(31.27±11.45)个月,两组患者总生存期的差异存在统计学差异。此外 CNTN-1高表达患者的无疾病生存期为(13.61±8.63)个月,低于低表达组患者(24.51±10.41)个月。多重变量分析表明CNTN-1表达量的高低是影响HCC患者总生存期和无疾病生存期的独立预后因素[28]。
在38例CNTN-1蛋白表达阳性的胃癌患者中,3年和5年生存率分别是46.1%和23.0 %,在29例表达阴性患者中则分别为76.7%和57.0%。Kaplan-Meier 生存曲线显示,胃癌患者的术后生存率与CNTN-1蛋白表达相关,具有显著统计学差异[29]。
综上所述,虽有多项研究证实CNTN-1蛋白与预后相关,不同的表达水平可能会影响患者生存期长短,但由于入组患者数量较少、观察时间较短、治疗方案选择不一致、肿瘤分期不同已经患者个体之间的差异性对患者生存期存在不可忽视的影响,可能存在一定的偏倚,仍有待进一步更大样本量的临床观察。另外,探讨CNTN-1蛋白与患者预后的关系也应增加关于年龄、淋巴转移、术后治疗方案等具体分组,以免造成因观察因素混杂造成的偏倚。同时,虽基础研究提示CNTN-1蛋白与肿瘤耐药性相关,但并无大样本队列研究观察肿瘤药物耐药时长与CNTN-1表达的数据,在该领域的临床观察亟待补充。
4 小结和展望
在人类多种恶性肿瘤中,CNTN-1都通过多种方式在肿瘤细胞的发生、发展、浸润和远处转移中发挥重要作用,且与疾病的分期与预后密切相关。目前研究提示CNTN-1蛋白在肿瘤细胞中的表达与否、表达强度和疾病的预后具有重要的评估和推测作用,且可能是潜在的生物标志物,因此,针对CNTN-1蛋白的持续研究是非常必要的。然而这种蛋白表达相关研究在恶性肿瘤的发生发展所起到的作用和相关作用机制尚不清楚且缺少相关研究证据。近年来一些关于CNTN-1与恶性肿瘤间关系的研究不断涌现,但基本上是停留在蛋白层面的描述性研究,深入阐明其发生发展分子机制的研究仍较匮乏。通过研究CNTN-1在多种恶性肿瘤中的发生、发展过程中所起到的作用及其相关分子机制,可能会为以后靶向治疗提供新的靶向治疗位点、为预后预测提供可靠指标。
利益冲突:所有作者均申明不存在利益冲突。
猜你喜欢
中国现代医生(2022年19期)2022-11-04
中国医学物理学杂志(2022年9期)2022-10-09
中国现代医生(2022年19期)2022-08-25
三农资讯半月报(2021年1期)2021-01-27
中国医药导报(2016年33期)2017-03-06
中国现代医生(2016年31期)2017-03-02
中国现代医生(2016年32期)2017-03-02
中学生物学(2016年10期)2016-11-19
现代养生·下半月(2015年8期)2015-11-16
销售与管理(2006年9期)2006-09-17
