
Targeted Genotyping of a Whole-Gene Repertoire by an Ultrahigh-Multiplex and Flexible HD-Marker Approach
2022-08-17 01:51PingpingLiuJiLvCenTinqiZhngXiowenHungZhihuiYngLinglingZhngbJingjieHudShiWngbZhenminBoc
Engineering 2022年6期
Pingping Liu, Ji Lv*, Cen M Tinqi Zhng Xiowen Hung Zhihui Yng Lingling Zhngb,Jingjie Hud, Shi Wngb,d, Zhenmin Boc,d,*
a Key Laboratory of Marine Genetics and Breeding and Sars-Fang Center, (Ministry of the People’s Republic of China) Ocean University of China, Qingdao 266003, China
b Laboratory for Marine Biology and Biotechnology, Pilot Qingdao National Laboratory for Marine Science and Technology, Qingdao 266237, China
c Laboratory of Tropical Marine Germplasm Resources and Breeding Engineering, Sanya Oceanographic Institution, Ocean University of China, Sanya 572000, China
d Laboratory for Marine Fisheries Science and Food Production Processes, Pilot Qingdao National Laboratory for Marine Science and Technology, Qingdao 266237, China
Keywords:HD-Marker Targeted genotyping Whole gene repertoire Non-model organism
ABSTRACT Targeted genotyping is an extremely powerful approach for the detection of known genetic variations that are biologically or clinically important. However,for non-model organisms,large-scale target genotyping in a cost-effective manner remains a major challenge. To address this issue, we present an ultrahigh-multiplex, in-solution probe array-based high-throughput diverse marker genotyping (HDMarker)approach that is capable of targeted genotyping of up to 86 000 loci,with coverage of the whole gene repertoire,in what is a 27-fold and six-fold multiplex increase in comparison with the conventional Illumina GoldenGate and original HD-Marker assays,respectively.We perform extensive analyses of various ultrahigh-multiplex levels of HD-Marker (30 k-plex, 56 k-plex, and 86 k-plex) and show the power and excellent performance of the proposed method with an extremely high capture rate(about 96%)and genotyping accuracy(about 96%).With great advantages in terms of cost(as low as 0.0006 USD per genotype) and high technical flexibility, HD-Marker is a highly efficient and powerful tool with broad application potential for genetic, ecological, and evolutionary studies of non-model organisms.
1. Introduction
The profiling of phenotype-related genetic variations lies at the heart of modern genetics. Genetic polymorphisms—particularly single-nucleotide polymorphisms (SNPs)—have been widely used in a variety of research applications in the fields of ecological,agricultural, and medical sciences [1–3]. More recently, with the advent of various high-throughput sequencing platforms,genome-wide SNPs have become readily attainable, providing unprecedented opportunities in numerous and diverse genomic applications [4]. Whole-genome sequencing enables researchers to identify and genotype SNPs at a complete resolution; however,it is still largely cost-prohibitive for the vast majority of organisms with moderate-sized or large genomes when dealing with a large number of samples (e.g., hundreds to thousands) [5]. Several reduced genome sequencing methods based on the use of restriction enzymes have been developed and are widely used [6], and can identify and genotype large-scale SNPs in a cost-effective manner(e.g.,restriction-site associated DNA(RAD)[7],genotyping-bysequencing (GBS) [8], and type IIB restriction-site associated DNA(2b-RAD) [9,10]). However, only the SNPs that are adjacent to restriction sites are sequenced and genotyped,making these methods more suitable for de novo marker discovery and genotyping.Gene-related markers that are located in or near the candidate genes of interest are more likely to be involved in phenotypic trait variation and are extremely valuable for the study of ecological,agricultural, and medical genetics [11,12]. With abundant ‘‘functional”marker resources having been accumulated,a diverse range of genetic research and applications are best achieved through target genotyping [13,14]. Although array-based genotyping platforms (e.g., the Illumina Infinium and Axiom Affymetrix platforms) are very powerful and are widely applied in human and model organisms[15–18],they are largely inaccessible to most non-model organisms due to the lack of inexpensive standardized commercial arrays [17,19]. Moreover, SNP arrays are fixed once established, which makes the adjustment of target loci difficult.Furthermore,fixed arrays developed in limited germplasm samples can sometimes suffer from inherent ascertainment bias [20].
Recent advances in sequencing technologies have accelerated the development of sequencing-based target-enrichment methods that promise to overcome some of the limitations of array-based platforms. To date, a variety of sequencing-based strategies for the genotyping of target loci have been reported,which can largely be classified into polymerase chain reaction (PCR)-based approaches and in-solution hybrid capture approaches [21,22].PCR-based approaches (e.g.,microdroplet PCR [23]and Ion Ampli-Seq[24])are amenable to automation that allows the processing of a large number of samples.However,major drawbacks include the dependency on specialized instruments(e.g.,RainStorm or Ion Proton systems) and potential primer competition in the highly multiplexed PCR reaction, which can lead to the generation of nonspecific amplification products [21]. The complexity of potential primer interactions in multiplex PCR presents the great obstacle to further multiplexity increase of amplicons(up to 20 000 SNPs in a single tube) [21,25].
In comparison, in-solution hybrid capture approaches (e.g.,NimbleGen and SureSelect), which can handle large amounts of target regions (especially for megabase-scale regions), are prominent target sequence capture methods[26–28].The target capacity of in-solution hybrid capture approaches usually ranges from 250 kb to 5 Mb of the captured region,so such approaches are capable of genotyping more than 50 000 loci [21,27,29]. Moreover, a very small amount of DNA (less than 10 ng) or even partially degraded DNA can be used, making in-solution hybrid capture approaches advantageous over array-based platforms [30,31].However, most of these approaches are more commonly used to detect variants across broad genomic regions of interest instead of examining specific loci of particular interest.[21,22,30].In addition,these approaches generally show a low target specificity that ranges from 40% to 60% [26]. Such a high degree of off-target sequencing may substantially impact data quality and adequate coverage across the intended targets,eventually leading to greater sequencing costs at high coverage [15,27,32–35].
To date, achieving the targeted genotyping of a whole gene repertoire in a cost-effective and flexible manner remains a major challenge, especially for non-model organisms. We previously reported a sequencing-based high-throughput diverse marker genotyping (HD-Marker) approach that relies on a high-density in-solution probe array and permits the targeted genotyping of up to 12 472 user-defined markers in a single-tube assay with high flexibility in the choice of multiplex levels and marker types [36].The methodology of HD-Marker is based on locus-specific probes(LSPs) that target the flanking sequences of the loci of interest and, through simple, highly multiplexed extension, ligation, and amplification steps, accomplish library construction for highthroughput target sequencing. HD-Marker combines the advantages of the high specificity and flexibility of GoldenGate technology with the cost-effectiveness of sequencing platforms. We have demonstrated the excellent performance of HD-Marker at multiplex levels of several hundred to 12 472 SNPs with an extremely high capture rate (over 98%) and genotyping accuracy (over 97%),which indicates that HD-Marker is a promising and attractive tool for large-scale targeted genotyping in non-model organisms.However, HD-Marker’s capacity for SNP multiplexity has not yet been fully explored, and the previously tested multiplexity (up to 12 000 loci) is still suboptimal for fulfilling large-scale targeted genotyping of all genes.In the present study,we demonstrate that the multiplex capacity of HD-Marker can reach the ultrahigh level of more than 86 000 loci, with an extremely high capture rate(about 96%) and genotyping accuracy (about 96%), along with a cost as low as 0.0006 USD per genotype. The ultrahighmultiplexing, high flexibility, and excellent performance of HDMarker makes it an ideal and powerful tool for accomplishing large-scale targeted genotyping in non-model organisms.
2. Methods
2.1. Probe design and preparation
High-quality(HQ)assayed SNPs were generated by the genome resequencing of 30 individuals of Yesso scallop (Patinopecten yessoensis(P.yessoensis))collected from diverse geographical locations in Liaoning Province in China. These individuals included samples from the Donggang,Zhuanghe,and Dachangshan populations,as well as two breeding varieties, Haida golden and Zhangzihong scallop, with six individuals per population or variety. We selected SNPs with a minor allele frequency between 0.2–0.5 for the 86 k-plex panel design, thereby representing common genetic variants among populations. Each SNP locus was targeted by two separate probes (LSP1 and LSP2). The principles used in the probe design basically followed those reported in our previous study[36]. In brief, there were two locus-specific probes that comprise unique flanking sequence and universal primers for a given locus.The flanking sequence refers to the upstream sequence spanning positions -22 to -1 and downstream sequence spanning positions +5 to +26, respectively (SNP coordinates representing zero).Probes with 40% to 60% GC content and 55 to 65 °C melting temperature were retained for further alignment.We removed the loci located in the regions that have multiple mapping results across the reference genome with two mismatches. The LSP1 and LSP2 sequences of all probes are provided in (Table S1 in Appendix A).The LSP1 and LSP2 were then combined with universal oligo application primers and specific restriction site sequence for Nt.AlwI,Nb.BsrDI,and Nt.BsmAI to generate 126 bp oligos.Oligonucleotide probes were from CustomArray, Inc. (USA), a commercial supplier of a cost-effective, high-throughput oligonucleotide synthesis method. These array-synthesized oligos were amplificated,digested and isolated by magnetic beads to obtain the LSP1 and LSP2 probes. Detailed steps are described as follows.
2.1.1. PCR amplification
The initial oligo pool was diluted 200 times, and then amplificated with 1.8 μmol∙L-1of biotinylated primers (Oligo_F and Oligo_R), 0.6 mmol∙L-1deoxynucleotide (dNTP) solution mix,1 × Phusion HF buffer, and 0.8 units Phusion high-fidelity DNA polymerase (NEB, USA) in a reaction volume of 60 μL. Two tubes of 60 μL volume of PCR reaction were prepared. The PCR was performed under the following thermal conditions:98°C for 30 s,followed by 24 cycles of 98 °C for 15 s, 60 °C for 10 s, and 72 °C for 15 s,and the final extension at 72°C for 5 min.PCR products were mixed and purified using a QIAquick PCR purification kit (Qiagen,Germany) and eluted with 32 μL of pure water.
2.1.2. Enzyme digestion
The purified products were then digested with the restriction enzymes to make the separated LSP1 and LSP2. Approximately 2 μg of product (about 20 μL) was initially digested by 3 μL of Nt.AlwI (NEB) in a 60 μL volume at 37 °C for 3 h, and then heat inactivated at 80 °C for 20 min. Next, 3 μL of Nb.BsrDI (NEB) was added to the tube. The mixture of total 63 μL reaction was incubated at 65 °C for 3 h, and then at 80 °C for 20 min. Finally,4 μL of Nt.BsmAI (NEB) was added to the tube and the mixture was incubated at 65 °C for 3 h, and then at 80 °C for 20 min.
2.1.3. Probe isolation by means of magnetic beads
The streptavidin magnetic beads were applied to separate of the biotin-labeled strand that is complementary to the target probe.Firstly,50 μL of streptavidin magnetic beads(NEB)was washed by 50 μL of washing buffer (0.5 mol∙L-1NaCl, 20 mmol∙L-1Tris-Cl,and 1 mmol∙L-1EDTA).Then,67 μL of digested product was added to the tube and mixed well using a pipette, while avoiding the generation of bubbles. The mixture was subsequently incubated at room temperature for 20 min. Denature the digested products mixture for 5 min at 95 °C and then quickly chilled them on ice for 5 min. A magnetic separation device was applied to pull the magnetic beads to the side of wells for easily supernatant removal.Aspirate the liquid into a new tube and left beads in the wells.Then purify the supernatant by using a Nucleotide Removal Kit(Qiagen).The isolated probe pool was eluted using 30 μL of elution buffer(10 mmol∙L-1Tris-Cl, pH 8.5), and was then ready for hybridization.
2.2. Library preparation and sequencing
2.2.1. Preparation of biotin-labeled genomic DNA
Adult individual P.yessoensis scallops were used for the evaluation of HD-Marker assays. Phenol/chloroform extraction method was applied to extract HQ DNA from the scallop adductor muscles[37]. Before hybridization, 3 μg of DNA samples was labeled with biotin by thermal coupling, following the protocols of PHOTOPROBE biotin labeling kit (Vector Labs, USA).
2.2.2. Hybridization
An optimized procedure was developed to achieve the effective hybridization of ultrahigh-multiplex probes. A total of 5–10 μL of biotin-labeled DNA was added to a tube containing 10 μL of magnetic beads,which had been washed twice with 50 μL of UltraHyb-Oligo hybridization buffer (Ambion, USA) in advance. After incubating the mixture at room temperature for 5 min, the magnet was applied and the supernatant was discarded. Then, approximately 15–30 μL probe mixtures were added to the tube, and UltraHyb-Oligo hybridization buffer (Ambion) was added to reach a total volume of 100 μL. All of the mixture was placed in a MyCycler thermal cycler (Bio-Rad, USA) by ramping the temperature from 70 °C to 30 °C over a period of about 8 h.
2.2.3. Extension and ligation
After hybridization, the magnetic beads were washed twice using washing buffer 1 (2 × salinesodium citrate (SSC) buffer,0.5% sodium dodecyl sulfate (SDS) buffer) and washing buffer 2(2 × SSC), respectively, to remove non-specific unbound probes.Gap filling and ligation were performed in a 25 μL reaction composed of 0.4–0.8 units Phusion high-fidelity DNA polymerase(NEB), 40–80 units Taq DNA ligase (NEB), 1 mmol∙L-1NAD (β-Nicotinamide adenine dinucleotide) (NEB), 0.1 mmol∙L-1dNTPs,and 1 × Phusion HF buffer. The reaction was then incubated at 45°C for 20 min.After gap filling and ligation,wash the beads with elution buffer(10 mmol∙L-1Tris-Cl,pH 8.5),and then resuspended in 35 μL of elution buffer. Finally, heat the mixture at 95 °C for 1 min to release the ligated products.
2.2.4. Library preparation and sequencing
HD-Marker sequencing libraries were constructed, following our previous protocol [36]. In brief, ligated products (about 30 μL) were amplified with 0.1 μmol∙L-1each of two universal PCR primers using 0.8 units of Phusion high-fidelity DNA polymerase (NEB) in a total volume of 50 μL. These PCR conditions were used: 26 cycles of 10 s at 98 °C, 20 s at 60 °C, and 10 s at 72 °C, followed by a final extension of 5 min at 72 °C. Check the amplified product (116 bp) on 8% polyacrylamide gel and cut the target band out of the gel.The gel-purified product was reamplified for seven cycles to introduce next-generation sequencing (NGS)platform-specific adaptor sequences and barcodes using the same PCR program. It was then purified using a QIAquick PCR purification kit(Qiagen)and eluted using 32 μL of pure water.The purified PCR product was quantified via Qubit and run on a Bioanalyzer(Agilent, USA) to check the library quality. The library was then subjected to Illumina HiSeq PE150 sequencing (Novogene, China).
2.3. Data processing and analysis
The forward reads (R1) of all the samples were first preprocessed to cut into 50 bp from the first base for subsequent analysis.Low quality reads that have ambiguous basecalls (N), long homopolymer regions (>10 bp) or excessive low-quality positions(>20% of positions with a quality score <10) were discarded. A set of about 50 bp sequences surrounding the target loci were extracted as reference. The remained high-quality reads were aligned to the reference using Burrows-Wheeler Alignment tool(BWA)[38].The output alignment files were sorted and converted into mpileup files using the SAMtools pipeline [39]. SNPs were genotyped by Varscan [40] with the parameters ‘‘--min-coverage 8 --min-reads2 2 --min-var-freq 0.01 --min-freq-for-hom 0.99 --p-value 99e-2”.Loci with a coverage greater than or equal to eight reads were used for reliable genotype calling.
To assess the genotyping accuracy of HD-Marker,genome resequencing was conducted using the same individual. The DNA libraries were constructed by using the Next-Ultra DNA Library Prep Kit for Illumina (NEB) in duplicate. The libraries were subjected to Illumina HiSeq X-Ten sequencing, with a total coverage of approximately 21×. The sequencing data were aligned to the P. yessoensis reference genome (GenBank accession no.GCA_002113885.2) using BWA [38]. Varscan [40] was used to genotype SNPs, with the parameters ‘‘--min-coverage 3 --minreads2 1 --min-var-freq 0.01 --min-freq-for-hom 0.99 --p-value 99e-2”. Consistent genotypes from replicate libraries were used to validate the HD-Marker genotypes.
The sequencing data from this study were submitted to the National Center for Biotechnology Information (NCBI) Sequence Read Archive†† https://www.ncbi.nlm.nih.gov/sraunder the accession numbers PRJNA669118 and PRJNA669126.
3. Results
3.1. SNP panel choice and library setup
We investigated the ultrahigh-multiplexing potential of HDMarker using the scallop P. yessoensis, which is one of the best molecularly characterized molluscs [41,42], with a HQ reference genome and abundant SNP resources [43–45]. High-quality assayed SNPs were generated by the genome resequencing of 30 scallop individuals collected from diverse geographical locations.In total,we obtained 2 044 646 SNPs that met the stringent criteria of the probe design. Three large panels (30 k-plex, 56 k-plex, and 86 k-plex) with a wide genomic distribution were selected from these HQ SNPs (Fig. 1). The majority of the SNPs in the three large panels were located in the genic regions, with 65.78% of the SNPs in the 30 k-plex, 71.81% of those in the 56 k-plex, and 70.08% of those in the 86 k-plex being located in the genic regions (Fig. S1(a) in Appendix A). These panels covered approximately 20 100 genes, including 87% of the Swissprot-annotated and 90% of the GO-annotated genes in the P.yessoensis genome[45],with approximately one SNP, two SNPs, and three SNPs per gene in the 30 kplex, 56 k-plex, and 86 k-plex, respectively (Fig. 1). Of the SNPs located in the genic regions, 52.30%–56.12% and 8.17%–12.24%were derived from the exonic and 3′-/5′-untranslated region(UTR) regions, respectively, whereas 32.06%–39.53% were derived from the intronic regions (Fig. S1(b) in Appendix A and Table 1).In order to compare genotyping results obtained by different multiplex levels, all SNP loci in a given multiplex level should be reserved in higher levels to maximize the numbers of loci shared between two or three plexes (e.g., the 86 k-plex contains all loci in the 30 k-plex and 56 k-plexes,and the 56 k-plex contains all loci in the 30 k-plex).In total,six HD-Marker libraries were prepared at three multiplex levels with two technical replicates per multiplex level for Illumina sequencing.
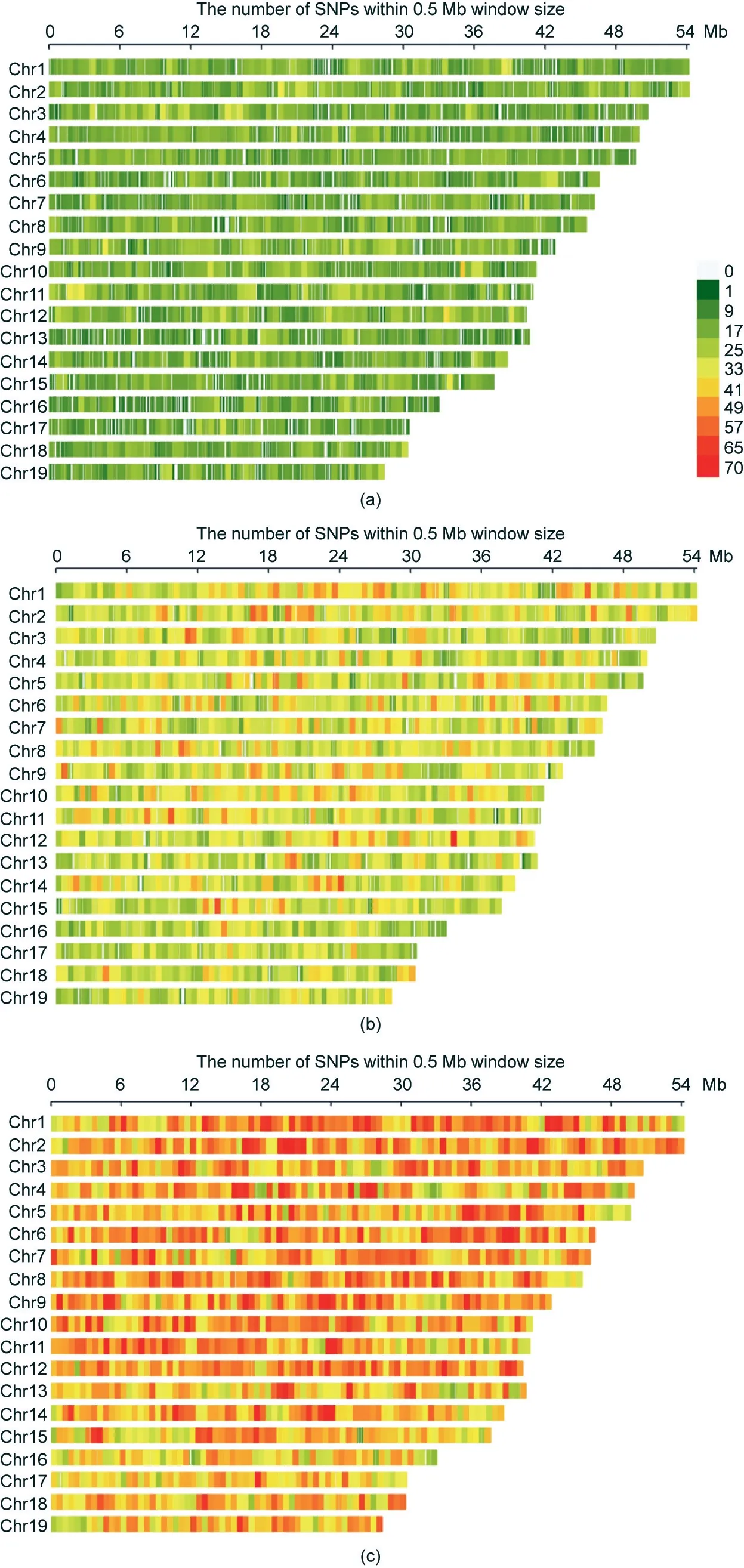
Fig. 1. Chromosomal distribution of SNP markers for three multiplex levels. (a) 30 k-plex; (b) 56 k-plex; (c) 86 k-plex.
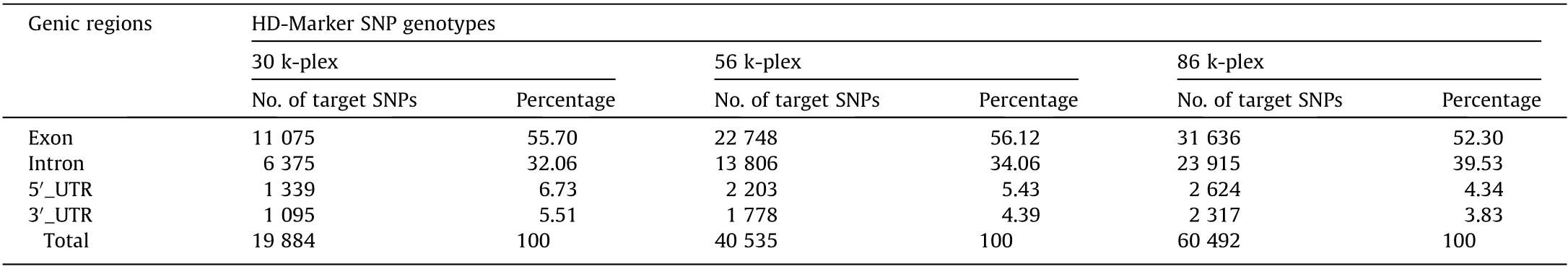
Table 1 Distribution of target SNPs in the genic regions of P. yessoensis.
The original protocol for 12 k-plex HD-Marker library preparation[36]is suboptimal for genotyping an ultrahigh number of loci(e.g., 86 k-plex), because it often results in low concentrations of prepared libraries that are sometimes insufficient for Illumina sequencing.To conquer this problem,we optimized the hybridization conditions by removing the unsuccessful biotin-labeled DNA using magnetic beads before hybridization and optimizing the ratio of probes and biotin-labeled DNA (i.e., 15–30 μL probe mixtures in contrast to 5–10 μL in the original protocol, and 500 ng of biotin-labeled DNA in contrast to 200 ng in the original protocol). The effectiveness of the library preparation (in comparison with the original protocol) was significantly improved, as demonstrated by the gel electrophoresis analysis (Fig. S2 in Appendix A).
3.2. Specificity, capture rate, and uniformity
Firstly, we examined the fraction of the sequencing reads that aligned with the target regions(i.e.,specificity),because this factor reflects the ability of the method to enrich appropriate targets and can greatly influence the cost(as more sequencing is required for a lower aligned fraction).Sequencing the six libraries produced more than 20 million, 33 million, and 49 million reads, respectively, for each technical replicate library in the 30 k-plex, 56 k-plex, and 86 k-plex, of which 98.53%–99.80% were retained as HQ reads for further analysis (Table 2). Approximately 81.24% of the HQ reads could be aligned to the target regions in the 30 k-plex, compared with 79.98%in the 56 k-plex,and 79.72%in the 86 k-plex(Table 2).The specificity in the 56 k-plex and 86 k-plex was only slightly lower (about 1%) compared with that in the 30 k-plex, indicating the high specificity of the HD-Marker assay in the three large panels.
Secondly, we examined the coverage of the target loci (i.e., the capture rate).The majority(96.65%–96.94%)of the target loci were detected with no noticeable difference in the three multiplex levels, showing the high capture rate of HD-Marker (Table 3 and Fig. 2). Sites detected in the replicate library exhibited high reproducibility,accounting for more than 97.57%in one replicate library and 97.64% in the other replicate library in the three large panels(Table 3). In addition, the locus detection was highly reproducible across the multiplex levels.The majority of detected loci(99.65%–99.71%; Table 4) in the 30 k-plex and 56 k-plex could be detected in higher multiplex levels, indicating highly repeatability across multiplex level.
Thirdly, we examined the uniformity of the coverage depth,because high uniformity indicates that fewer sequencing reads are needed to generate adequate coverage of the target loci for the downstream analysis, making the sequencing more cost-effective. The sequencing depth for the commonly detectedsites at different multiplex levels have high Pearson correlation(0.92 between technical replicates and 0.91–0.92 between multiplex levels)(Fig.3).The sequencing coverage of the target loci varied within 2–4 orders of magnitude, with 94.63%, 93.49%, and 93.24% of the loci falling within a 100-fold range (Fig. 4), as revealed by quantification of the capture uniformity of the three large SNP panels. The high uniformity of the capture loci seemed to be unaffected by the GC content, with Pearson’s r ranging from 0.060 to 0.098(Fig.S3 in Appendix A),indicating that the gradient cooling program we adopted ensures that most of the probes correctly anneal to the targeted sites.
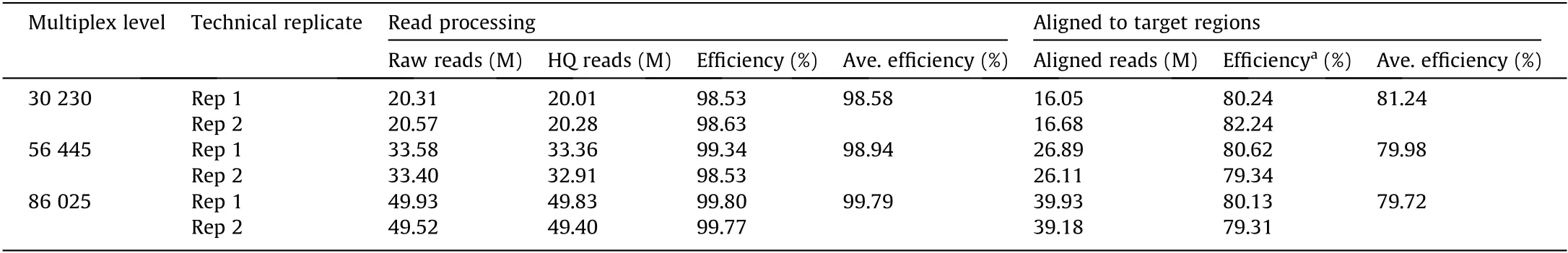
Table 2 Illumina data processing and alignment to target regions.
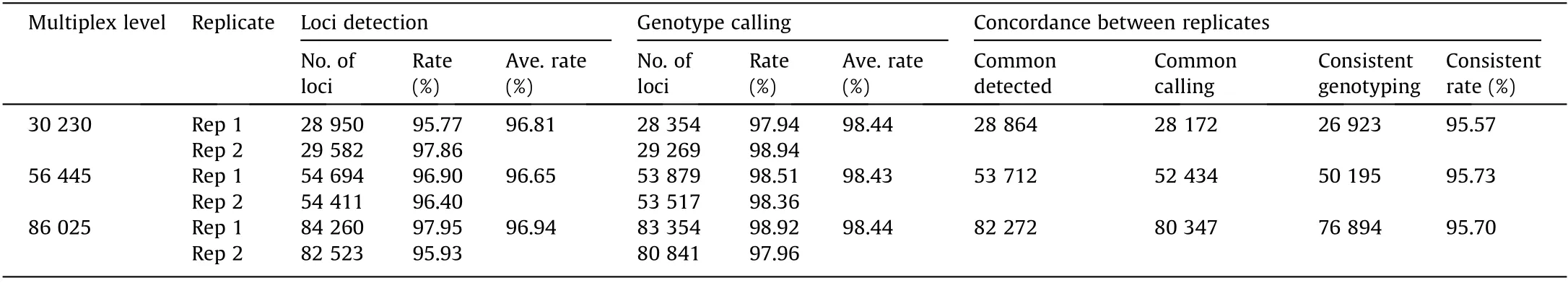
Table 3 Summary of loci detection, genotype calling, and concordance between replicates.
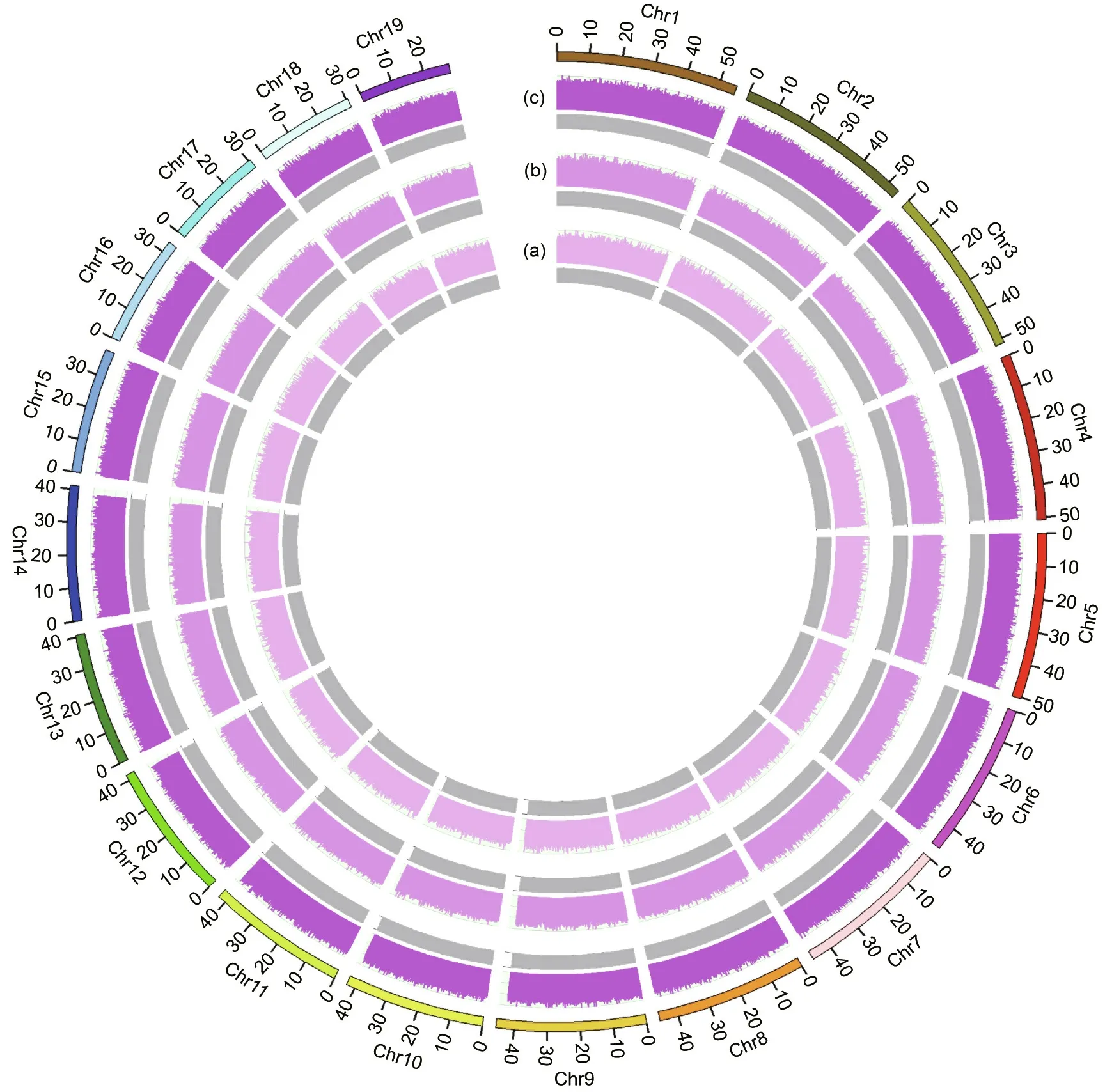
Fig.2. Sequencing coverage of target SNP markers for three multiplex levels.An extremely high capture rate(about 96%–98%)and even sequencing coverage across loci were observed for all multiplex levels. (a) 30 k-plex; (b) 56 k-plex; (c) 86 k-plex.
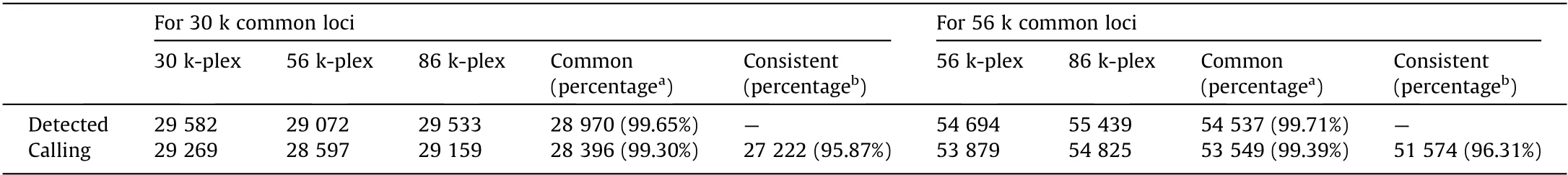
Table 4 Genotyping performance of common target SNPs across multiplex levels.
3.3. Genotype calling and accuracy
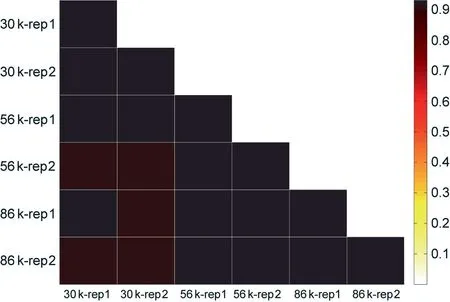
Fig. 3. Pearson correlation heatmap showed that the sequencing coverage of commonly detected loci is highly correlated across technical replicates (r = 0.92)and multiplex levels (from r = 0.91 to r = 0.92). rep: replicate.
We further examined the number of sequencing reads that align to each target locus, because the precision of the SNP genotyping depend on minimum coverage depth. Over 98% of the detected sites were covered by greater than or equal to eight reads(98.44%,98.43%,and 98.44%for the 30 k-plex,56 k-plex,and 86 kplex, respectively), so we set eight as the coverage threshold for genotype calling (Table 3). A high level of genotype calling rates(97.94%–98.94%) was observed across two technical replications for each of three multiplex levels (Table 3). We then evaluated genotyping accuracy by measuring the concordance of our genotype result in three different aspects. Firstly, for the comparison between genotype calls derived from the replicate libraries,approximately 95.57%–95.73% of the common calling loci showed the same genotyping result between two replicate libraries in three multiplex levels (Table 3). Secondly, for the comparison of the genotype calls of common loci across different multiples levels,we observed a genotype concordance of 95.87%–96.31% between the three multiplex levels (Table 4). Lastly, whole genome resequencing was conducted based on the same assayed individual to validate genotyping accuracy. All three multiplexes achieved a genotype accuracy of greater than 96% (Table 5), indicating the high genotyping accuracy of HD-Marker across all multiplex levels.
Heterozygous sites are generally more difficult to be correctly genotyped than homozygous sites. When considering heterozygous sites and homozygous sites separately,we found that the concordance rate at positions found to be heterozygous was more than 96.29% for all multiplex levels (Table 5). Notably, the distribution of allelic sampling closely matched the expectations both within and across multiplex levels,converging to 0.5 for heterozygous loci and to 1 for homozygous loci (Fig. 5).
3.4. Rarefaction and cost analysis
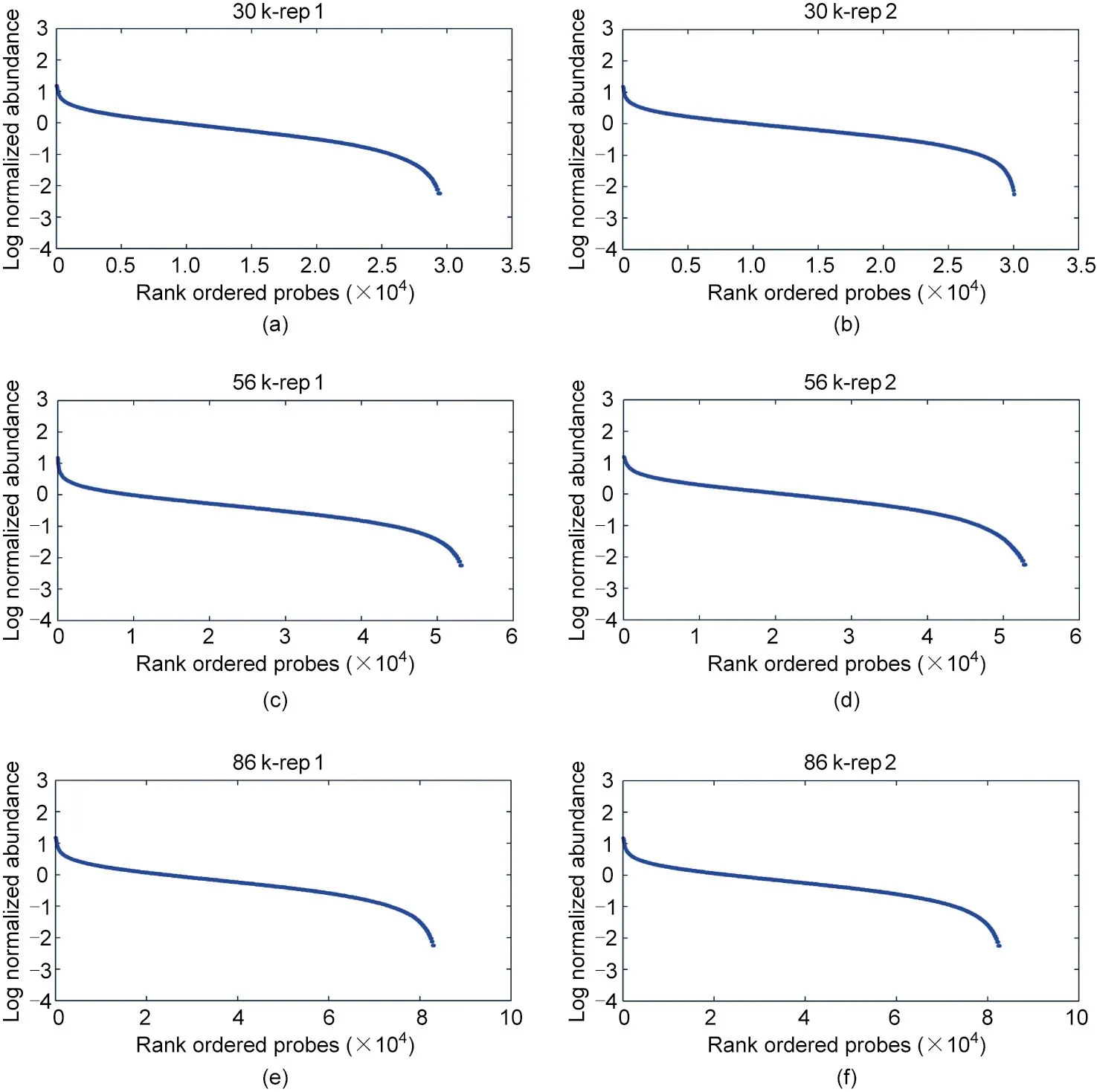
Fig.4. Illustration of capture uniformity for different multiplex levels. The log(base 10)values of the relative sequencing depth of each target locus were calculated, sorted and plotted.The capture uniformity varies within 2–4 orders of magnitude for(a,b)30 k-plex,(c,d)56 k-plex and(e,f)86 k-plex,with 93.24%–94.63%of loci falling within in a 100-fold range.
Table 5 Genotype validation by genome resequencing.
Fig.5. Performance of allelic sampling for three multiplex levels.(a,b)30 k-plex;(c,d)56 k-plex;(e,f)86 k-plex.The distribution of allelic sampling largely converges to 0.5 for (a, c, e) heterozygous loci, but converges to 1 for (b, d, f) homozygous loci.
To determine the optimal amount of sequencing to achieve cost-effective targeted genotyping, we combined dataset of two replicate libraries to perform rarefaction analysis for each multiplex level. For all three large SNP panels, we observed an initial sharp rise in the detected rate, calling rate, and genotyping accuracy as the amount of sequence data increased. This was followed by a plateau, during which very little gain was obtained with additional sequencing data (Fig. 6). When the amount of sequencing reads reached 5 million, 10 million, and 13.5 million for the 30 k-plex, 56 k-plex, and 86 k-plex, respectively, the locus detection rate was saturated,with 95.8%–96.5%of the detected loci being genotyped at the corresponding depths (Fig. 6). At the optimal amount of sequencing,genotyping accuracy of 96.40%,96.01%,and 96.15% could be achieved for 30 k-plex, 56 k-plex, and 86 kplex, respectively. Rarefaction analysis allows the calculation of detection rate and genotyping accuracy for a given amount of sequencing,so we could estimate the minimum sequencing depth needed to genotype the required target loci.Our analysis facilitated the identification of an ideal balance among the number of target loci, genotyping accuracy, and cost.
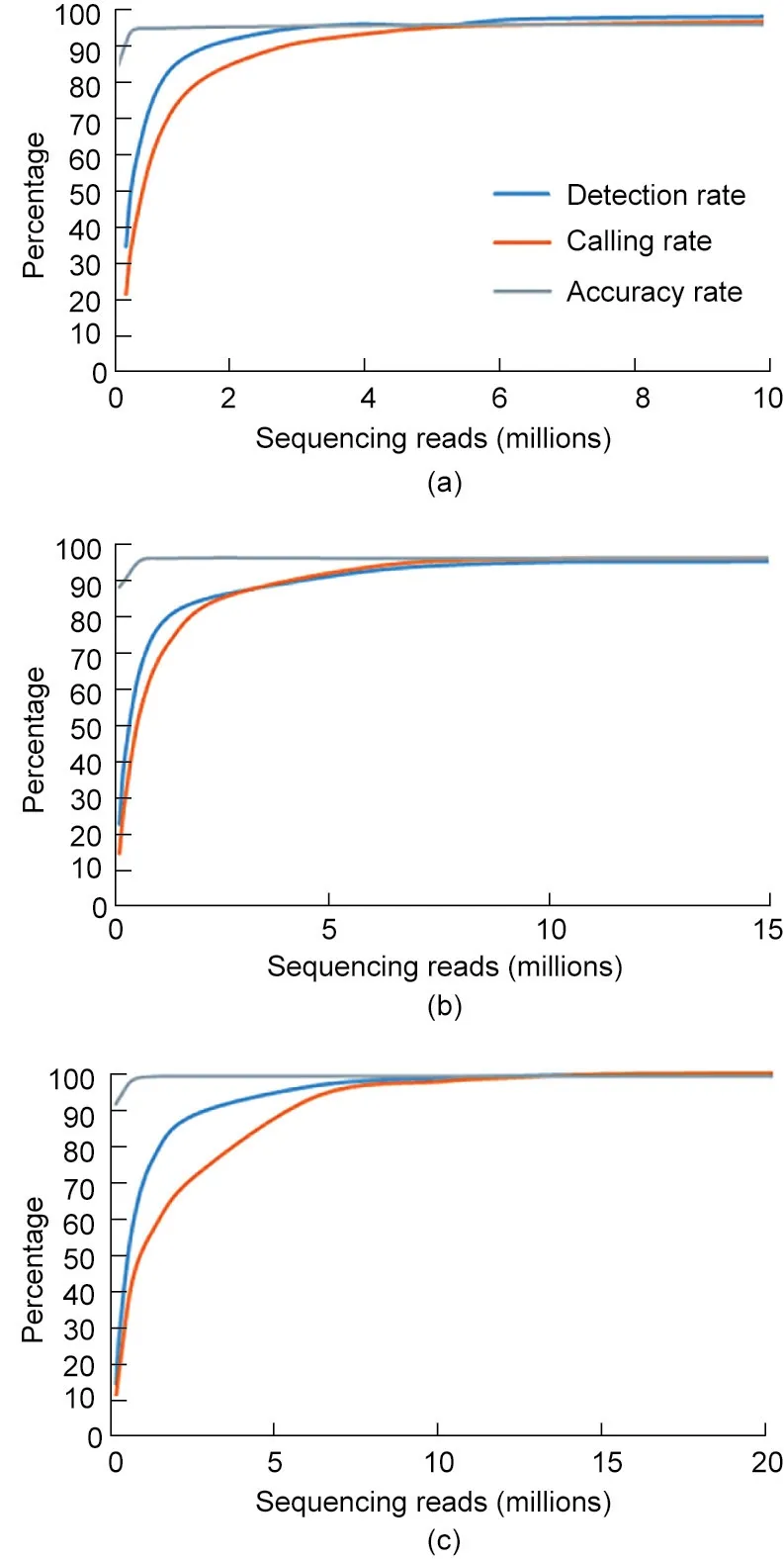
Fig. 6. Rarefaction curves for the detection rate, calling rate and accuracy rate at different sequencing scales. For (a) 30 k-plex, (b) 56 k-plex, and (c) 86 k-plex, loci detection is saturated at 5 million, 10 million, and 13.5 million reads, respectively,and a genotyping accuracy of 96.40%, 96.01%, and 96.15%, respectively, can be achieved at optimal sequencing depths.
We further estimated the genotyping cost(including the cost of probe synthesis, library preparation, and NGS sequencing) under different numbers of samples for each multiplex level.The optimal sequencing reads were estimated based on the rarefaction analysis.HD-Marker genotyping is very economical, with per-sample costs of 29.4–92.1 USD, 44.1–106.7 USD, and 58.5–121.2 USD for multiplex levels of 30 k-plex, 56 k-plex, and 86 k-plex, respectively(Table 6). Moreover, the cost per sample or per genotype become increasingly cheaper as the number of samples increased. For example,for 86 k-plex,the cost per sample for a scale of 100 samples was 121.20 USD,whereas the cost per sample was 64.23 USD when handling 1000 samples (Table 6), as larger sample size contributes to the reduction of the probes costs per sample.The genotyping cost per locus can reach as low as 0.0006 USD at 86 k-plex.
4. Discussion
Targeted genotyping is highly effective and has the power to genotype user-defined genetic variations that are biologically or clinically important. Yet, in the case of non-model organisms,genotyping of large amounts of target loci at low-cost(e.g.,for tens of thousands to hundreds of thousands of loci)remains a challenge.Current targeted genotyping approaches suffer from several inherent limitations,such as genotyping of only up to thousands of loci for the PCR-based method (e.g., microdroplet PCR and AmpliSeq)[21,25]; high expense and time consumption when building custom arrays (array-based genotyping; e.g., Affymetrix arrays)[5,46]; and the targeting of wide range of genomic regions rather than specific loci (e.g., Agilent SureSelect) [21,22]. Illumina’s GoldenGate has been proposed as a promising tool for genotyping a large set of genomic loci and it is well known for its high SNP multiplexity and high flexibility of SNP selection [47–50]. However, original GoldenGate assay is bead array-based and involves the use of fluorescently labeled primers which require special instrument to detect. Our previous study demonstrated that this methodology, when switched to the NGS platform, allows more than 12 000 loci to be genotyped simultaneously in a single tube[36].
In the current study, we presented an ultrahigh-multiplex HDMarker approach that permits the targeted genotyping of up to 86 000 loci and thus enables the coverage of the whole gene repertoire, in what is a 27-fold and six-fold increase over conventional Illumina GoldenGate and original HD-Marker assays, respectively.To achieve this goal,we optimized the original protocol by removing unsuccessful biotin-labeled DNA using magnetic beads before hybridization and adjusting the number of probes and biotinlabeled DNA for the effective hybridization of 86 000 loci. In this work, the 86 000 SNP panel mostly represented common genetic variants (based on the resequencing data of 30 individuals with diverse geographical backgrounds), and the resequencing of a larger number of individuals may be necessary if both common and rare variants need to be targeted. The extensive analyses in terms of specificity, capture rate, uniformity and genotype reproducibility and accuracy verified the robustness and excellent performance of HD-Marker at various multiplex(30 k-plex,56 k-plex,and 86 kplex).Target SNPs can be further increased by mixing multiple different sets of 86 k-plex probes which is comparable to that of conventional microarrays capable of genotyping for hundreds of thousands to millions loci.High-density SNP assays would provide the sufficient ability to cover population wide linkage disequilibrium that is necessary for genome-wide association studies [51].Moreover, the use of high-density panel would increase theprediction accuracy in genomic selection[52].With such technical improvement, HD-Marker exhibits several advantages (e.g.,ultrahigh-multiplexing, high efficiency, and flexibility) that can overcome the inherent limitations of other techniques, such as a relatively lower loci multiplexity(only up to thousands)for microdroplet PCR and AmpliSeq; high expense, time consumption, and low application flexibility for developing custom fixed microarrays;and the targeting of broad genomic regions of interest rather than specific loci for the currently widely used in-solution hybrid capture approaches. In addition, HD-Marker exhibits much higher specificity (79.72%–81.24%) in comparison with the relatively low specificity(about 52%–57%)of widely used in-solution hybrid capture approaches [26]. It also provides a great advantage in cost, at 29–121 USD per sample for the 30 k-plex to 86 k-plex, which is a cost reduction of approximately 40%–60%in comparison with commonly used targeted genotyping methods [53]. Furthermore, HDMarker exhibits high genotyping accuracy in comparison with the current gold-standard of genome resequencing (with >96%genotyping consistency for all multiplex levels).
Table 6 Genotyping costs comparisons between different multiplex levels and sample scales.
HD-Marker offers a scalable multiplex and flexible approach for high-throughput targeted genotyping especially in non-model organisms. Unlike array-based method, the entire process of HDMarker assay can be easily adopted in ordinary laboratories without requirement of any expensive, specialized instruments.It provides researchers with increased power and flexibility regarding the marker number and type to meet specific research purposes.Furthermore,researchers can take the cost and number of loci into account in order to select the appropriate level.Small panels of SNP markers are a better choice for parentage assignment and the determination of relatedness in breeding programs, whereas large panels can be used when aiming to generate linkage maps, estimate trait heritability, or perform quantitative trait locus (QTL)mapping and genomic selection [54]. Our HD-Marker approach, if combined with the imputation-based strategy[55,56],allows a significant increase in loci number without additional cost and is therefore much more cost-effective.For genomic selection,a recent study has shown that using the top 500–2000 SNPs (derived from genome-wide association analysis) provides comparable or even better prediction accuracy than using all SNPs[57].At this marker level, a cost of below 10 USD per sample would be fully possible when adopting the HD-Marker approach. These are promising directions and worthy of further exploration. We envision that HD-Marker will become an attractive tool with broad application potential in genetic, ecological, and evolutionary studies of nonmodel organisms in the near future.
Acknowledgments
We acknowledge the grant support from National Natural Science Foundation of China (32130107, 32002446 and 32102778), Project of Sanya Yazhouwan Science and Technology City Management Foundation (SKJC-KJ-2019KY01), China Agriculture Research System of MOF and MARA,and Taishan Scholar Project Fund of Shandong Province of China.
Compliance with ethics guidelines
An early version of HD-Marker has been granted a Chinese patent (ZL201310549040.7).
Pingping Liu, Jia Lv, Cen Ma, Tianqi Zhang, Xiaowen Huang,Zhihui Yang, Lingling Zhang, Jingjie Hu, Shi Wang, and Zhenmin Bao declare that they have no other conflict of interest or financial conflicts to disclose.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.eng.2021.07.027.

- Engineering的其它文章
- Electric Air Taxis Create Megadeal Buzz
- Tissue Engineering and Regulatory Science
- Factors Predicting Progression to Severe COVID-19: A Competing Risk Survival Analysis of 1753 Patients in Community Isolation in Wuhan,China
- A Vaccine Based on the Receptor-Binding Domain of the Spike Protein Expressed in Glycoengineered Pichia pastoris Targeting SARS-CoV-2 Stimulates Neutralizing and Protective Antibody Responses
- Fabrication and Applications of Multi-Fluidic Electrospinning Multi-Structure Hollow and Core–Shell Nanofibers
- Past and Future Changes in Climate and Water Resources in the Lancang–Mekong River Basin: Current Understanding and Future Research Directions