
Tissue Engineering and Regulatory Science
2022-08-17 01:51PengZhoWenoLiuJixinTinXinliShiXiosongGuAntoniosMikos
Engineering 2022年6期
Peng Zho, Weno Liu, Jixin Tin, Xinli Shi, Xiosong Gu, Antonios G. Mikos
a Center for Medical Device Evaluation, National Medical Products Administration, Beijing 100081, China
b Jiangsu Key Laboratory of Neuroregeneration, Nantong University, Nantong 226001, China
c Departments of Bioengineering, Chemical and Biomolecular Engineering, Rice University, Houston, TX 77251, USA
Tissue engineering has successfully evolved from its original concept [1] into medical products with a rapid pace of development and a multi-billion dollar market [2]. Compared with traditional medical products, tissue-engineered medical products(TEMPs) have distinct characteristics that provide unique benefits for the repair and regeneration of damaged or diseased tissues or organs [1,2]. For example, as pioneer TEMPs with living cells that have been approved by the US Food and Drug Administration(FDA),Apligraf products have been used in clinics for the treatment of venous leg ulcers since 1998 and were later expanded to treat diabetic foot ulcers. The first autologous cellularized scaffold for the repair of knee cartilage defects (MACITM) received FDA approval in the United States in 2016, and represents a breakthrough that differs from traditional orthopedic treatments of osteoarthritis by means of joint replacements. In 2015, the Pharmaceuticals and Medical Devices Agency (PMDA) approved the first regenerative medicine product in Japan, HeartSheet, which is a cell sheet technology-based product for patients with serious heart failure. The National Medical Products Administration of China(NMPA)has also approved several TEMPs such as a cellularized skin product, xenografts developed from pig corneas, and a peripheral nerve graft composed of conduits of chitosan, chitin,gelatin, and poly(lactic-co-glycolic acid) (PLGA) fibers. In addition,a few TEMPs under review at the Center for Medical Device Evaluation (CMDE) have received the NMPA’s designation of innovative medical devices (IMDs) [3,4].
Despite their advantages, TEMPs present significant challenges in terms of their regulatory evaluation, review, and approval by various regulatory agencies across the world [5,6]. The process from basic research on a TEMP to its successful clinical translation goes through multiple phases, including: designation, classification, and regulation;preclinical evaluations;manufacturing; quality system; clinical evaluation; post-market evaluation.
1. Designation, classification, and regulation
TEMPs must be appropriately and clearly designated,classified,and regulated as medical devices, combination devices, drugs, or biological products.The designation,which is based on the primary mode of action of the specific TEMP, can be complicated but will determine the TEMP’s regulatory pathways. For example, the MACI product mentioned earlier is designated and regulated as a biological product in the United States. Another TEMP named INFUSE, which is a bone graft that comprises recombinant human bone morphogenetic protein-2 applied to an absorbable collagen sponge carrier, is designated and regulated as a combination product in the United States. If a TEMP is designated as a medical device, depending on its risk and relevant control procedures, it may be classified and regulated as a Class III or II device.
2. Preclinical evaluations
Preclinical research and evaluations of TEMPs may need new tools, methods, standards, or guidance documents, which can strongly impact their regulatory and commercialization processes.Scientifically challenging examples include—but are not limited to—the following:
∙How to evaluate compatibility issues between living tissues and TEMPs that consist of biomaterial scaffolds, bioactive factors, and live cells;
∙How to trace and evaluate the fate of live cells in scaffolds;
∙How to ensure the quality of final TEMPs that cannot be terminally sterilized;
∙How to assess the quality of regenerated tissues during and after TEMP remodeling;
∙ How to evaluate the synergistic effects of biodegradable scaffolds of TEMPs on biocompatibility, cell fates, and regenerated tissue.
3. Manufacturing
Manufacturing is widely recognized as one of the biggest hurdles for the commercialization of TEMPs. The manufacturing processes of TEMPs are more complex than those of traditional medical products and thus present significant challenges for scale-up production and quality control. Compared with traditional medical products,TEMPs need well-designed raw materials,along with potentially living cells and bioactive molecules.Therefore, a significant investment in advanced infrastructures and manufacturing technologies is needed for safe and effective production that will ensure end-product quality. Technical challenges include—but are not limited to—the following:
∙How to set up regulated manufacturing facilities that are designed only for TEMPs;
∙How to identify and ensure the quality of manufacturing equipment for TEMPs;
∙How to perform process validations for TEMPs and identify
the key process parameters.
4. Quality system
A robust quality management system (QMS) via either current good manufacturing practice(cGMP)or quality systems regulation(QSR)is required to ensure the safety,efficacy,quality,and performance of TEMPs. The unique characteristics, benefits, and risks of the TEMPs should be considered and regulated under a QMS via their specific risk management processes. Quality challenges for TEMPs may include—but are not limited to—the following:
∙For TEMPs that are combination products, how to establish a quality system that meets the requirements of both medical devices and drugs, or biological products;
∙How to set up specification and testing standards for raw materials, including live cells and/or biological factors;
∙How to develop in-line non-destructive testing methods and standards of critical quality attributes to accurately assess TEMPs;
∙How to develop and determine the criteria for the batch qualification of TEMPs.
5. Clinical evaluation
For the clinical translation of TEMPs, clinical evaluations,including clinical trials regulated by good clinical practices(GCPs),should be conducted with appropriate selections of indications,control groups, and primary and secondary endpoints. More specifically, the following issues should be considered:
∙For TEMPs that are combination products, how to coordinate the different requirements for the clinical trials of medical devices and drugs/biological products;
∙How to statistically design clinical trials based on indications;
∙How to identify gold standards or control groups for TEMPs;
∙How to apply evidence-based research approaches to design clinical trials and evaluate clinical data.
6. Post-market evaluation
Lastly, post-market surveillance of TEMPs would require strong scientific understanding of their properties and modes of action, even after successful regulatory approval and market commercialization processes. So-called ‘‘real-world research”collects real-world data and translates them into real-world evidence.
All of the above regulatory and technical challenges must be resolved through scientific and evidence-based approaches. As a result, regulatory science will be an important part of the tissue engineering process from basic research to commercialization,and new forms of regulatory science will have to be initiated and developed for this context.
Compared with the science that originates from curiosity and interest, regulatory science is driven by the vision, mission, and needs of regulatory authorities in order to best serve citizens [7].Regulatory science was not initially established for medical products. In the 1970s, the newly established the United States Environmental Protection Agency (EPA) encountered a difficult situation in which public decisions had to be made with insufficient scientific evidence[8].Inspired by this and other similar situations,Dr.A.Alan Moghissi of the EPA founded the Institute for Regulatory Science, a non-profit organization that conducted scientific research‘‘at the interface between science and the regulatory system,” in the spring of 1985 [8]. Dr. Mitsuru Uchiyama, who was working at the National Institutes of Health Sciences in Japan at that time, is regarded as the first scholar to propose the concept of regulatory science in Japan (1987) [8]. In 1990, Professor Sheila Jasanoff of Harvard University gave an analytical and rigorous description of the term ‘‘regulatory science” in her book titled
The fifth branch: science advisers as policymakers. As described in Jasanoff’s book, regulatory science is different from policy science and can be further extended to pharmaceuticals [8]. According to Jasanoff,‘‘regulatory science is that body of scientific and technical knowledge which serves regulatory decision making” [8]. It was not until the beginning of the 21st century that the concept of regulatory science was adopted by regulatory agencies such as the FDA; since then, it has developed into a multidisciplinary and cross-functional scientific field for the evaluation of the safety and effectiveness of medical products [9,10].
Regulatory science presents great opportunities, with new approaches, standards, and tools, to help in the evaluation of the safety, efficacy, quality, and performance of TEMPs as well as that of their innovations and fast-to-market translations [9,10] (Fig. 1).As early as 2010, the FDA started regulatory science programs to promote relevant scientific practices and fields in order to realize its regulatory missions and vision better. Since the approval of the 21st Century Cures Act in the United States in December 2016, the FDA has established new expedited product development programs for regenerative medicine advanced therapy(RMAT), which, as described in Section 3033 of the 21st Century Cures Act, includes a broad range of medical products such as TEMPs,cell therapy,human cell and tissue products,or any combination products using such therapies or products [11,12].
In November 2017,the FDA announced a comprehensive regenerative medicine policy framework to encourage innovation and provide efficient access to breakthrough yet safe and effective medical products. To further promote the regulatory science program for RMATs including TEMPs, the FDA also developed a guidance document on how to develop and use standards. In addition,it contracted with Nexight Group and with standards coordinating bodies (SCBs) such as the US National Institute of Standards and Technology (NIST), the International Standards Organization(ISO), and the American Society for Testing and Materials (ASTM)to coordinate community efforts toward the development of standards for RMATs [13]. Recently, the FDA’s Center for Devices and Radiological Health (CDRH)’s Office of Science and Engineering Laboratories (OSEL) published a variety of regulatory science tools that were developed to help with the assessment of innovative medical products, including TEMPs. Innovative TEMPs such as implantable and bioengineered human acellular vessels (HAVs;Humacyl)have benefited from the development of the FDA’s regulatory science programs through designations of RMATs and fasttrack review processes [12,14].
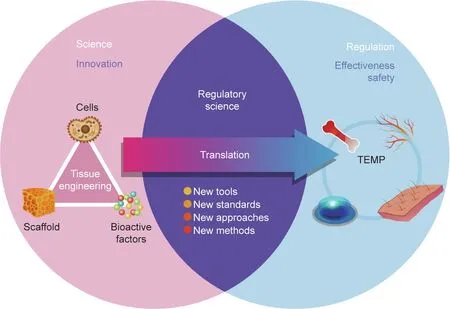
Fig. 1. A representative illustration of the overlap of science, regulatory science, and regulation, and of the translation from tissue engineering concepts to TEMP.
The global effort to advance regulatory science is also promoting the development of innovative medical products such as TEMPs in Europe and Japan.TEMPs are identified as one class of advanced therapy medicinal products (ATMPs) by the European Medicines Agency (EMA). The top strategic goal of EMA regulatory science is to‘‘catalyze the integration of science and technology in medicines’development;”furthermore,one of the core recommendations for this top strategic goal is ‘‘to support translation of ATMPs into patient treatments” [15]. EMA considers that ATMPs, including TEMPs, have great potential to both address unmet clinical needs and provide a wide range of treatments that are not available through traditional therapies. EMA has proposed multiple underlying actions to strengthen its core recommendations, one of which is to ‘‘provide assistance with early planning, method development and clinical evaluation” [15]. EMA’s Committee for Advanced Therapies (CAT) further published a reflection paper on clinical aspects related to TEMPs, which provides various considerations including claims, study design, and clinical safety and efficacy endpoints. In parallel to the FDA, Japan’s PMDA started its regulatory science activities around 2010,which led to the establishment of the PMDA Center for Regulatory Science in 2018 to further enhance product reviewing and safety measures. A new category of ‘‘regenerative medical products,”which includes TEMPs, was added to the Japanese Pharmaceutical Affairs Law, whose name was changed to the Pharmaceutical and Medical Device (PMD) Act, resulting in the conditional approval of Japan’s first regenerative medicine product, HeartSheet [16].
The NMPA has also launched regulatory science programs. In August 2018, the NMPA held the Seminar on Medical Device Regulatory Science, which was a landmark for Chinese regulatory science programs and a starting point for a series of subsequent regulatory science activities. In April 2019, the NMPA launched its Regulatory Science Action Plan(RSAP),which focuses on the five themes of innovation,quality, efficiency,system, and capability to promote the innovation of regulatory concepts and mechanisms,and to accelerate the progress of the medical product industry from ‘‘big” to ‘‘strong.” The RSAP identified three key tasks:①establishing scientific research bases for regulatory science;②launching key regulatory science projects; and ③developing new tools, standards, methods, and approaches for regulatory evaluation, review, approval, and supervision. Since the launch of the RSAP,a total of 12 regulatory science research bases have been established at universities and institutions across the country,and 117 key research laboratories have been designated by the NMPA.The research bases and labs provide a solid foundation and strong support for the development of regulatory science in China.
‘‘Combination products” and ‘‘cells and gene therapy products”were among the first batch of nine key regulatory science projects initiated by the NMPA in April 2019. The TEMPs Project is in the second batch of key projects of the RSAP, which was launched by the NMPA in July 2021. These three projects are all related to regenerative medicine products. One of the research objectives of the TEMPs Project is to investigate regulatory approaches for TEMPs, which include principles and considerations for the classification of TEMPs,the requirements of the quality system,and preclinical and clinical evaluation requirements. The other research objectives are to establish a safety and efficacy evaluation system for TEMPs with new tools, standards, and methods, which may include evaluating the effects of scaffolds on cell safety and functions in TEMPs.
Taken together, medical products are directly related to the health and safety of everyone in society. Innovative technologies present excellent market opportunities as well as regulatory challenges. Scientific evidence generated by new tools, standards,approaches, and methods is greatly needed [16] to evaluate the safety and efficacy of medical products,especially those developed from novel technologies such as tissue engineering and regenerative medicine. The global development of regulatory science for medical products such as TEMPs, with the engagement of academic, industrial, healthcare, and regulatory communities, not only ensures the safety and efficacy of medical products, but can also improve the efficiency of regulatory decision-making and product market entry. In this way, a balance of meeting clinical needs, encouraging innovations, and protecting public health can be achieved. Thus, public health can be promoted to the greatest extent.
Acknowledgments
The authors would like to thank the Center for Medical Device Evaluation (CMDE) of the National Medical Products Administration (NMPA) of China, as well as the NMPA’s Regulatory Science Action Plan (RSAP).

- Engineering的其它文章
- Electric Air Taxis Create Megadeal Buzz
- Factors Predicting Progression to Severe COVID-19: A Competing Risk Survival Analysis of 1753 Patients in Community Isolation in Wuhan,China
- A Vaccine Based on the Receptor-Binding Domain of the Spike Protein Expressed in Glycoengineered Pichia pastoris Targeting SARS-CoV-2 Stimulates Neutralizing and Protective Antibody Responses
- Fabrication and Applications of Multi-Fluidic Electrospinning Multi-Structure Hollow and Core–Shell Nanofibers
- Past and Future Changes in Climate and Water Resources in the Lancang–Mekong River Basin: Current Understanding and Future Research Directions
- Mass Transfer,Gas Holdup,and Kinetic Models of Batch and Continuous Fermentation in a Novel Rectangular Dynamic Membrane Airlift Bioreactor